

R如何读取不同类型的单细胞测序数据(10X、h5ad、RDS、loom)

我们拿到的单细胞测序数据的结果可能会有多种不同的类型,下面是几种不同类型单细胞测序数据的读取方法。
1、首先是读取经典的10X单细胞测序数据
10X单细胞测序数据经过cell ranger处理后会得到三个结果文件:matrix.mtx、barcodes.tsv 和genes.tsv。

matrix.mtx 即count矩阵
barcodes.tsv即样本的名称,也就是每个细胞的名称
genes.tsv即基因名称。
将这个三个文件放入同一个文件夹,打开R,将含有这三个文件的文件夹设置为工作路径。
1.1 准备工作
#加载Seurat包
> library(Seurat)
Attaching SeuratObject

Seurat为经典的单细胞分析工具,可以官方学习:
#查看一下工作路径
> getwd()
[1] "/media/xxx/F/scdata"#将工作路径赋值data_dir,方便后续使用
> data_dir <- '/media/xxx/F/scdata'# 查看工作路径文件夹下文件,证实路径无问题
> list.files(data_dir)
[1] "barcodes.tsv" "genes.tsv"
[3] "matrix.mtx" 1.2 正式读取
# Read10X命令读取三个文件,得到一个带行名(基因名)及列名(细胞名)的count的矩阵
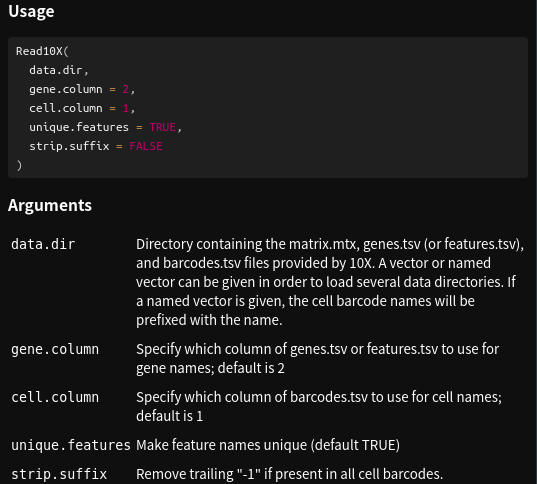
> count_matrix <- Read10X(data.dir = data_dir, gene.column = 1)# 查看一下Read10X函数使用帮助
> ?Read10X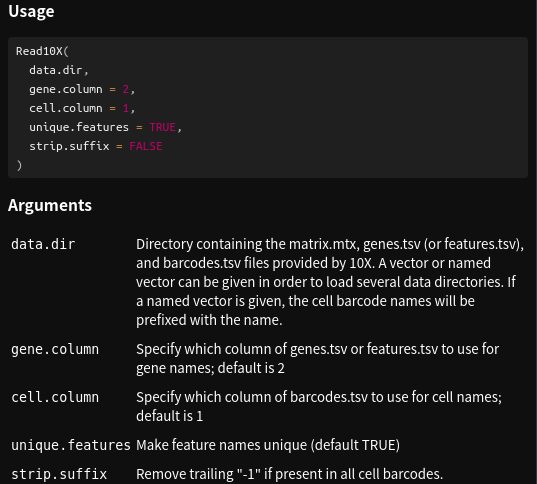
# 创建Seurat对象
> seurat_object = CreateSeuratObject(counts = count_matrix,
min.cells = 0,
min.features = 0,
project = "seurat_object")
# 可以查看一下CreateSeuratObject函数帮助文档
> ?CreateSeuratObjectcounts = count_matrix,将前面得到的count矩阵作为目标矩阵
如设置 min.cells = 3 ,表示某个基因至少要在3个细胞表达,不达标的基因将去除。
如设置 min.features = 100 ,表示某个细胞至少检测到有100个基因表达,不达标细胞将去除。
读取完毕,后续进行其他分析。
2、读取h5ad格式数据
2.1 首先安装转换格式需要的SeuratDisk包
# GitHub安装SeuratDisk包
> remotes::install_github("mojaveazure/seurat-disk")# 加载SeuratDisk包:
> library(SeuratDisk)2.2 转换格式,加载为Seurat对象
# 先将h5ad格式文件转换为 h5Seurat格式文件
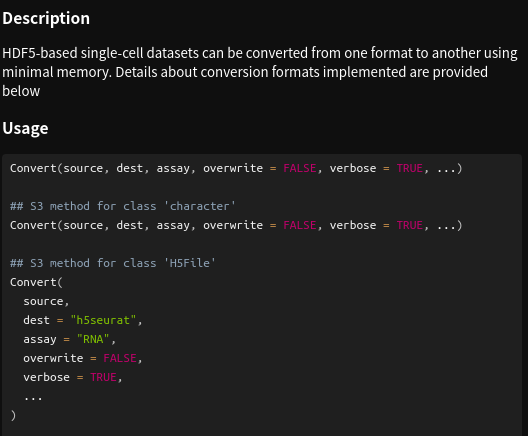
> Convert("xxxxx.h5ad", dest="h5seurat",
assay = "RNA",
overwrite=F)# 可以查看Convert函数帮助文档
> ?Convert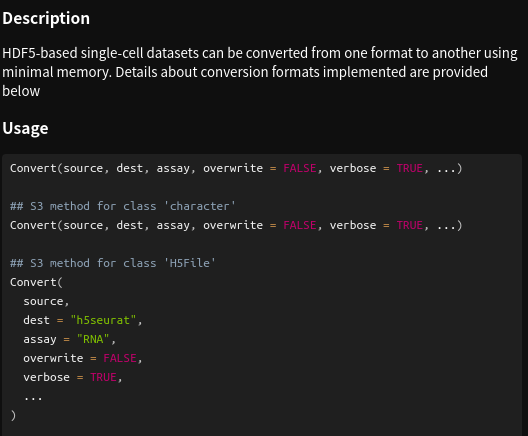
# 加载h5Seurat格式文件为Seurat对象
> seurat_object <- LoadH5Seurat("xxxxx.h5seurat")#需要的化升级Seurat对象为新版本Seurat对象
> library(Seurat)
> seurat_object = UpdateSeuratObject(seurat_object)
3、读取RDS格式数据
RDS后缀格式文件可以直接readRDS读取
##读取RDS文件为Seurat对象
> Seurat_object <- readRDS("xxxxxx.RDS")#需要的化升级Seurat对象为新版本Seurat对象
> library(Seurat)
> seurat_object = UpdateSeuratObject(seurat_object)
4、读取loom格式数据
需要用到loomR,有详细的官方指导:
4.1 安装loomR,连接loom对象
# 先安装devtools
> install.packages("devtools")# 用devtools安装hdf5r和loomR
> devtools::install_github(repo = "hhoeflin/hdf5r")
> devtools::install_github(repo = "mojaveazure/loomR", ref = "develop")# 加载loomR
> library(loomR)#连接( Connect) 到loom 文件
> lfile <- connect(filename = "xxxxxx.loom", mode = "r+")#查看loom 文件
> lfile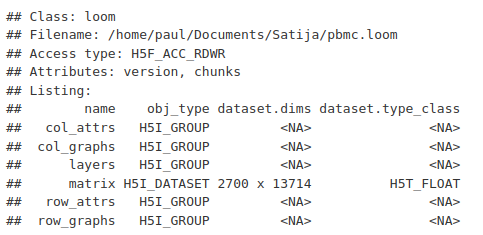
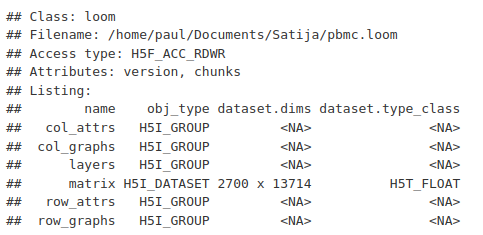
A
loom
object is a container for six sub-objects: one dataset (
matrix
) and five groups (
layers
,
row_attrs
,
col_attrs
,
row_graphs
, and
col_graphs
)。
4.2 转换loon文件为Seurat对象
没找到直接转换的方法,看官方指导的意思好像可以直接把loom文件当seurat文件来用:

可以如下手动转换:
> library(Seurat)
> library(SeuratDisk)#4.2.1 提取矩阵matrix
> matrix=lfile[["matrix"]][,] #提取loom文件的matrix#看一下矩阵大小
> dim(matrix)
[1] 10535 27998#发现那个matrix的gene和barcode颠倒了,换过来才符合seurat对象中的矩阵,行为基因,列为barcode
> matrix=t(matrix)
> dim(matrix)
[1] 27998 10535# 4.2.2 提取基因名和细胞名
> gene = lfile$row.attrs$Gene[]
> barcode = lfile$col.attrs$CellID[]
#查看一下长度
> length(gene)
[1] 27998
> length(barcode)
[1] 10535# 4.2.3 把基因名和细胞名加入矩阵
> colnames(matrix)= barcode