


变异信息定位染色体


确定基因组染色体长度信息
在进行变异信息定位到染色体上之前,需要先有染色体的长度信息才能进行定位,可以使用 TBtools或者samtools faidx
之前先写过推文了 染色体长度计算


变异位点信息文件准备
首先使用的是VCFtools过滤后的结果文件,其中包含变异存在的 染色体、位点及突变类型信息
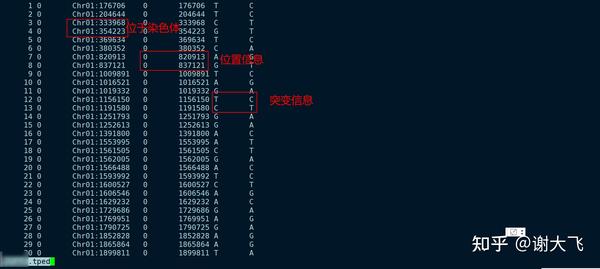
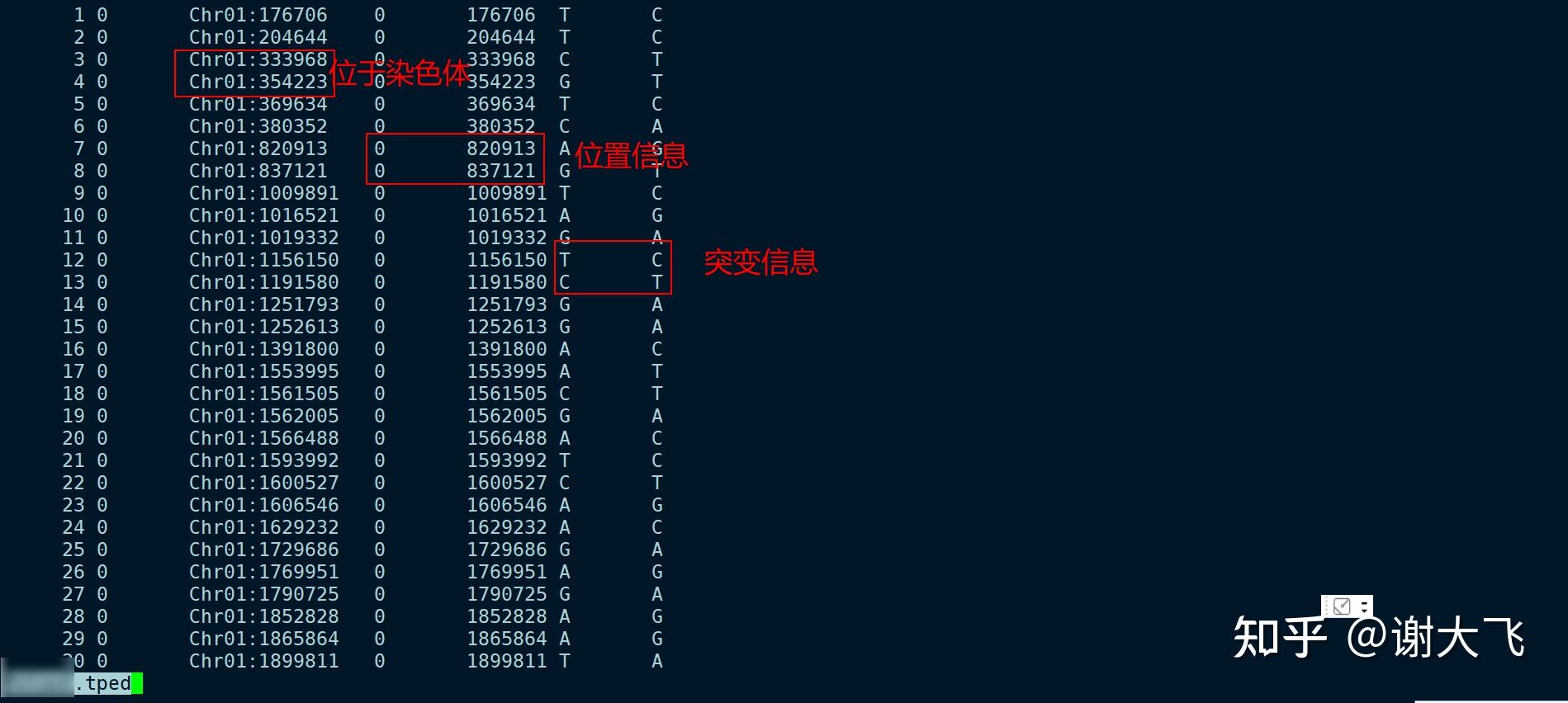
操作流程是: 服务器下截取结果文件中染色体标记以及位置信息生成txt格式的文件之后,放出来用Rstudio中使用RIdeogram实现定位到染色体上
1. 截取变异位点信息
因为使用 VCFtools 进行过滤之后的结果文件中包含的全部信息不能直接用于定位,所以需要截取 染色体及位点信息即可
cat XXX.tped | tr ":" "\t" | cut -f 2,4,5 >> XXX.txt
#tr ":" "\t" 将染色体位置信息后的:转换为tab
#cut -f 2,4,5 >> XXX.txt 截取需要的信息生成txt文件
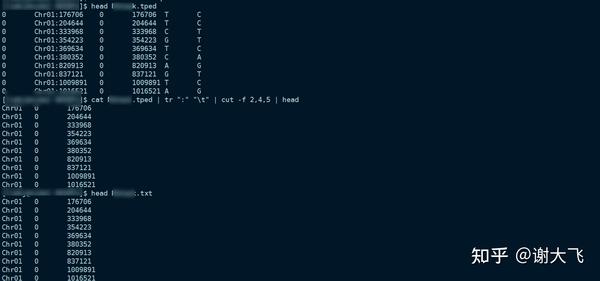
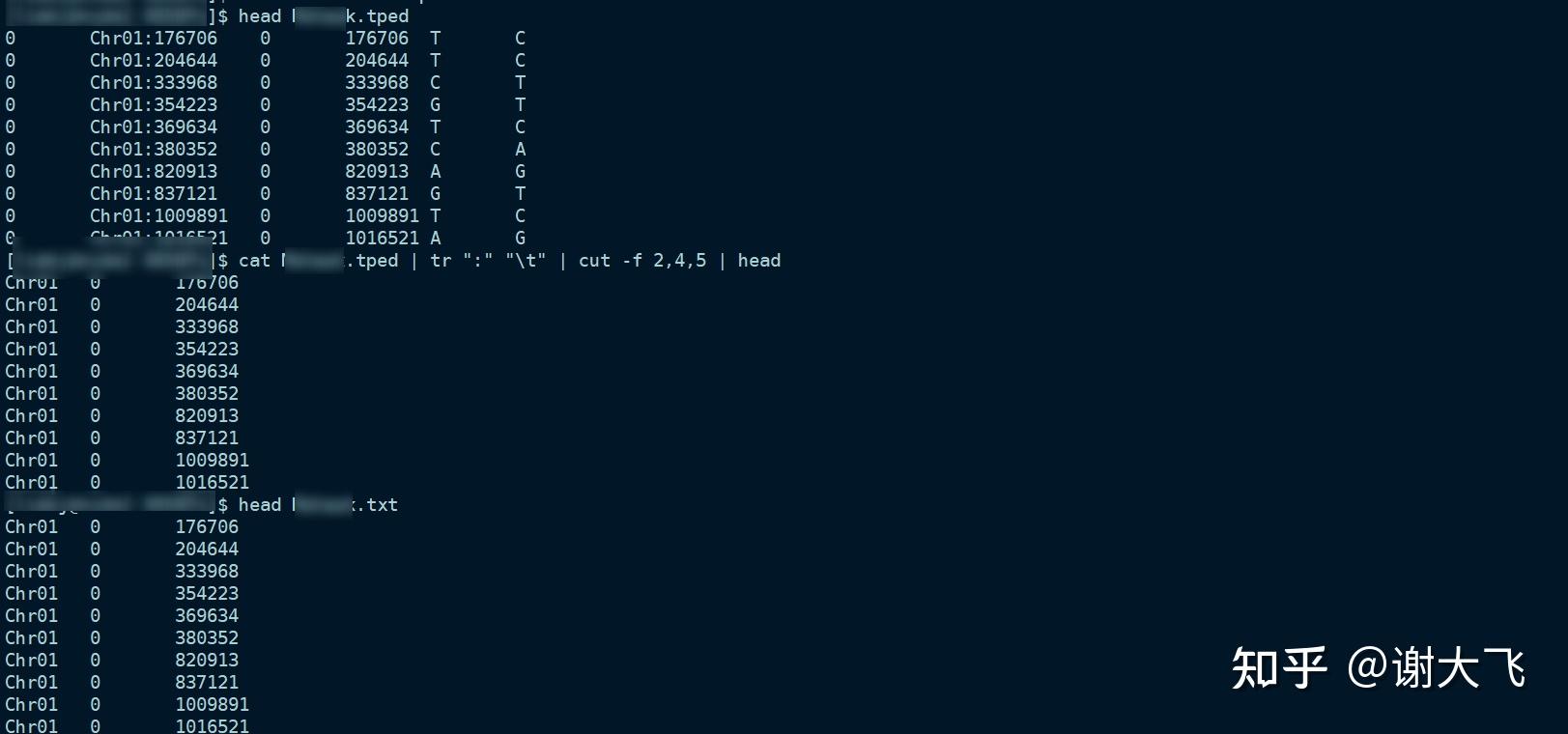
2. 将文件传输到本地电脑
我用的连接服务器的软件是 Xshell ,所以用来的传输文件的是配套的 Xftp
简单的窗口拖动文件即可
变异信息定位染色体
用到的R包是** RIdeogram **
(之前找教程的时候发现 TBtools 也是可以实现染色体定位的,但是我失败了,教程: TBtools | 全基因组特征标记可视化 - 太简单 )
1. 下载安装RIdeogram
install.packages('RIdeogram')
library(RIdeogram)
2.加载需要的示例数据
data("human_karyotype",package = "RIdeogram")
data("gene_density",package = "RIdeogram")
3. 导入自己的数据
导入染色体长度文件
#读取染色体长度文件
XXX_karyotype <- read.table("XXX_Chr_len.txt",sep = "\t",header = T,stringsAsFactors = F)
#使用软件提供的示例数据进行格式整理
data <- human_karyotype[,1:3]
data <- data[1:16,]
data$Chr <- XXX_karyotype$Chr
data$End <- XXX_karyotype$End
导入变异位点信息文件
#导入变异位点信息文件
XXX_density <- read.table("XXX.txt",sep = "\t",header = T,stringsAsFactors = F)
#使用示例数据进行列名更替