


WGCNA分析流程详解


前言
全文两万多字,建议收藏再看。
全文两万多字,建议收藏再看。
全文两万多字,建议收藏再看。
原文链接在此:
WGCNA,全称为weighted gene co-expression network analysis,即权重基因共表达网络分析。它是一种分析多个样本基因表达模式的分析方法,可将表达模式相似的基因进行聚类,并分析模块与特定性状或表型之间的关联关系,在研究表型性状与基因关联分析等方面的研究中被广泛应用。
什么是WGCNA?最简单的解释就是样本之间的各个基因是否存在共同表达的模式,例如基因A和基因B是否在某一个阶段中存在相同的表达模式——两者同时上调表达,还是同时下调表达。这个方法就是利用这样思路将样本中基因表达进行分析,探究基因间是否具有共表达的现象,并且根据一定的数值给某一团共表达的基因划分成一个模块,这样聚在一起的不同的团的基因就划分为不同的模块。例如关于调控花青素合成的基因可能就会聚类在同一个模块里面,关于调控叶绿素合成则可能会聚类在另一个模块里面。但是,WGCNA的分析还不止于此,它还可以利用这些模块和表型数据进行聚类,找到这个模块中的核心基因(权重较高的一些基因),也就是hubgene。
小编之前一直想学会WGCNA分析,但是网上的教程比较零散,而且方法不是很全面,所以一直耽误了比较久的时间。后来,小编直接看这个R包的事例操作,一步一步来分析,最后弄懂的。这个事例非常好,大家可以根据这个例子来操作几次,基本能掌握分析的流程了,不过是全英的版本的,可能对英语不好的伙伴不太友好,但是可以手动翻译呀。链接在下面:
https://horvath.genetics.ucla.edu/html/CoexpressionNetwork/Rpackages/WGCNA/Tutorials/为了更好上手,我把分析流程用中文整理了一遍,并且重点说了一下一些注意事项,所以不想看原教程的可以看我整理的流程。
WGCNA分析流程主要有以下几步:
- 数据输入与清洗(筛选)
- 网络构建与模块检测(有三种方法可以实现)
- 模块与表型数据关联并识别重要基因
- 网络交互分析(GO注释等)
- WGCNA网络可视化
- 导出网络互作数据(Cytoscape)
数据下载和准备,下载网址如下:
https://horvath.genetics.ucla.edu/html/CoexpressionNetwork/Rpackages/WGCNA/Tutorials/1. 数据输入、清洗和预处理
把数据中的缺失值、离群值、冗余数据数据去除
1.1 载入数据
设定R语言的工作路径
getwd("E:/Rdata/WGCNA")
workingDir = "E:/Rdata/WGCNA"
setwd(workingDir)
library(WGCNA) #加载WGCNA包
options(stringsAsFactors = FALSE) #开启多线程
femData = read.csv("LiverFemale3600.csv") #载入基因表达量数据
##这份*.csv文件是示例中的的3600个基因的表达数据
dim(femData)
names(femData)
#行为基因,列为不同样本的基因表达量或其他信息提取出表达量的数据 ,删去不需要的数据重新生成矩阵
datExpr0 = as.data.frame(t(femData[, -c(1:8)])) #提取加转置
names(datExpr0) = femData$substanceBXH #基因名字
rownames(datExpr0) = names(femData)[-c(1:8)] #样品名字
datExpr0 就是一个以每行为样本,一列为一个基因的数据框1.2 检查缺失值和识别离群值(异常值)
gsg = goodSamplesGenes(datExpr0, verbose = 3);
gsg$allOK如果gsg$allOK的结果为TRUE,证明没有缺失值,可以直接下一步。如果为FALSE,则需要用以下函数进行删除缺失值。
if (!gsg$allOK)
# Optionally, print the gene and sample names that were removed:
if (sum(!gsg$goodGenes)>0)
printFlush(paste("Removing genes:", paste(names(datExpr0)[!gsg$goodGenes], collapse = ", ")));
if (sum(!gsg$goodSamples)>0)
printFlush(paste("Removing samples:", paste(rownames(datExpr0)[!gsg$goodSamples], collapse = ", ")));
# Remove the offending genes and samples from the data:
datExpr0 = datExpr0[gsg$goodSamples, gsg$goodGenes]
}注:
在这一步之后,一般会有一个筛选基因的过程。因为电脑的配置问题,一般来说运行几万个基因的表达量来构建TOM矩阵和邻接矩阵是非常吃力的,我试过把全部的基因都用来构建网络,但是构建矩阵的过程非常费时间,而且很难能运行成功。所以在完成剔除异常值之后,我们还需要进一步挑选基因。至于怎么筛选基因要看自己的目的,粗暴一点的可以按表达量为前5000的进行提取,通常用5000个基因进行分析。(具体讨论可见WGCNA分析进阶版常见问题整理)
聚类所有样本,观察是否有离群值或异常值。
sampleTree = hclust(dist(datExpr0), method = "average")
sizeGrWindow(12,9) #视图
par(cex = 0.6);
par(mar = c(0,4,2,0))
plot(sampleTree, main = "Sample clustering to detect outliers", sub="", xlab="", cex.lab = 1.5,
cex.axis = 1.5, cex.main = 2)
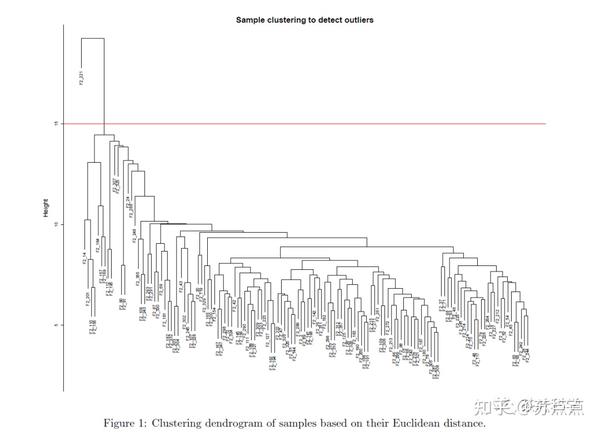
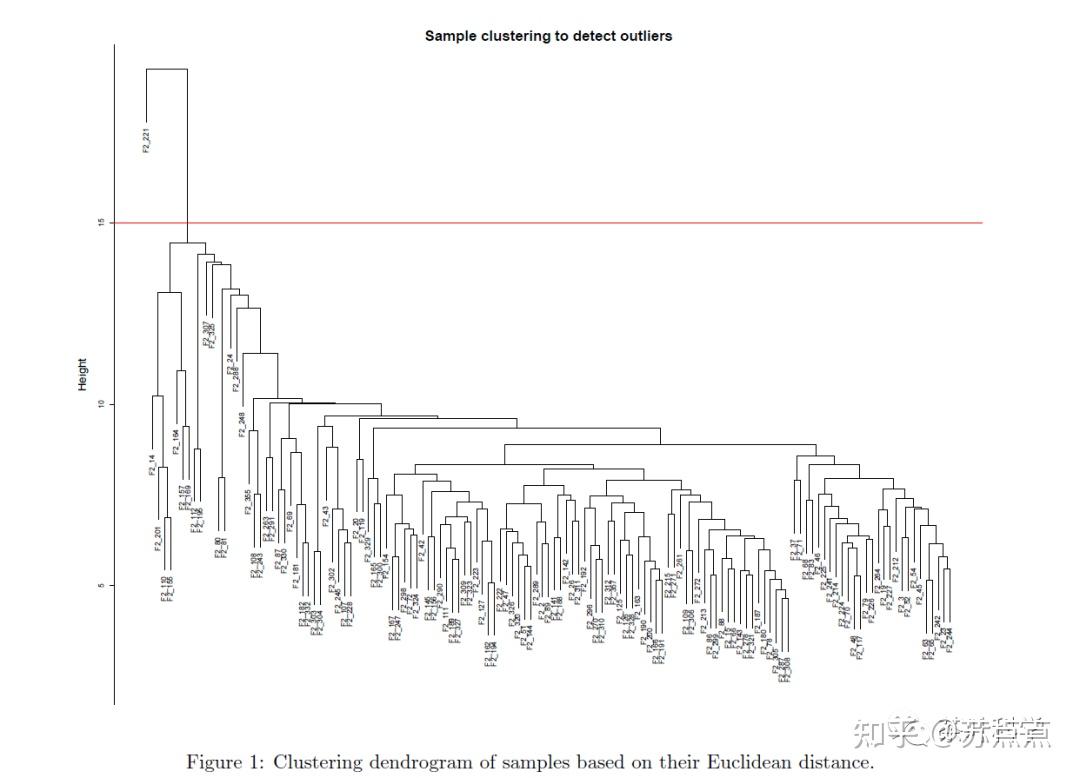
如果有离群值,则要删去离群的样本,如果没有则跳过下一步。
删除离群样本
abline(h = 15, col = "red") #划定需要剪切的枝长
clust = cutreeStatic(sampleTree, cutHeight = 15, minSize = 10)
这时候会从高度为15这里横切,把离群样本分开
table(clust)
keepSamples = (clust==1) #保留非离群(clust==1)的样本
datExpr = datExpr0[keepSamples, ] #去除离群值后的数据
nGenes = ncol(datExpr)
nSamples = nrow(datExpr)1.3 载入表型数据
traitData = read.csv("ClinicalTraits.csv");
dim(traitData) #每行是一个样本,每列是一种信息
names(traitData)
删除我们不需要的数据
allTraits = traitData[, -c(31, 16)];
allTraits = allTraits[, c(2, 11:36) ]; #只保留数值型数据
dim(allTraits)
names(allTraits)因为表型数据和基因表达量数据中并不是全部信息都能匹配上,例如表型数据中的样本可能在基因表达量数据中不存在,这时候需要将两者进行匹配,找共有的数据才能分析。
将临床表征数据和表达数据进行匹配(用样本名字进行匹配)
femaleSamples = rownames(datExpr);
traitRows = match(femaleSamples, allTraits$Mice);
datTraits = allTraits[traitRows, -1];
rownames(datTraits) = allTraits[traitRows, 1];
collectGarbage()可视化表型数据与基因表达量数据的联系,重构样本聚类树
sampleTree2 = hclust(dist(datExpr), method = "average")
traitColors = numbers2colors(datTraits, signed = FALSE) #用颜色代表关联度
plotDendroAndColors(sampleTree2, traitColors,
groupLabels = names(datTraits),
main = "Sample dendrogram and trait heatmap")图片结果解释了临床数据和基因表达量的关联程度,保存数据。
save(datExpr, datTraits, file = "FemaleLiver-01-dataInput.RData")
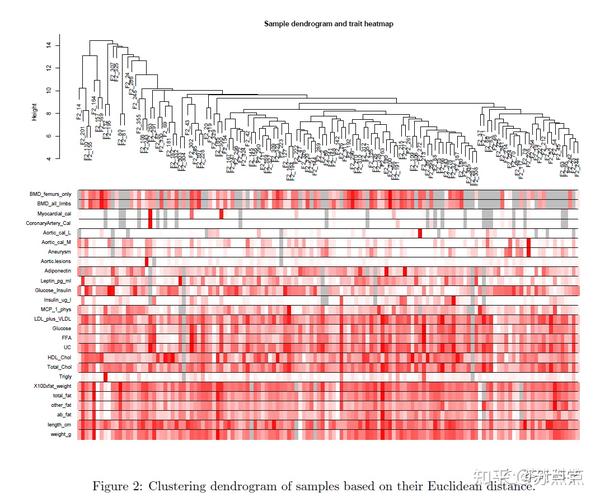
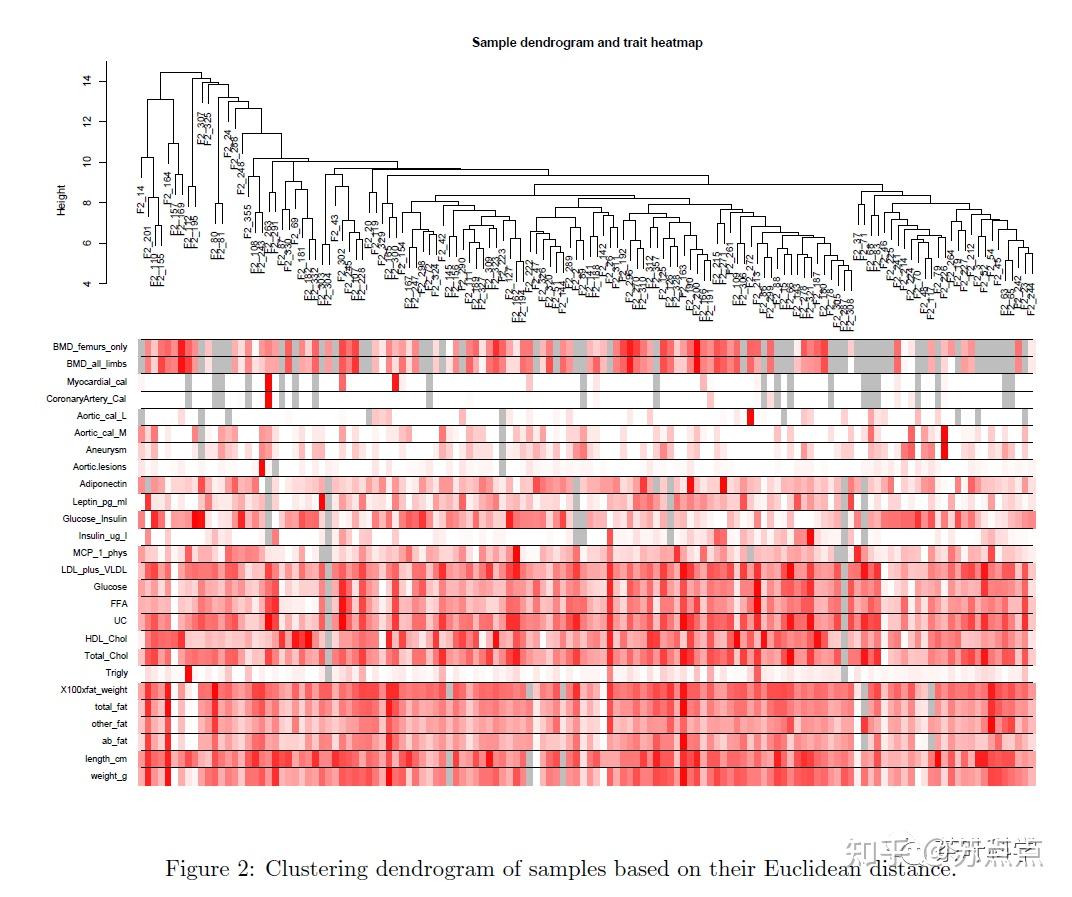
颜色越深,代表这个表型数据与这个样本的基因表达量关系越密切。
2. 构建表达网络
构建表达网络是WGCNA分析中最为关键的一步,是否构建成功、是否构建正确对后期模块的划分和关联表型数据筛选核心基因至关重要。
挑选软阈值是构建网络拓扑分析的关键,选择软阈值是基于近无尺度拓扑标准的。很多人会因为无法挑选合适的软阈值而卡在了这一步,不能往下走。其次就是构建TOM矩阵或者邻接矩阵的时候运行大数据无法成功。
除此之外,构建表达网络后划分模块的方法有三种,分别是 一步法、逐步法和分步法 ,不同的方法各有优缺点,要根据自己的需求选择。这三种方法的主要区别是:
- 一步法:适合处理较少的数据量,方便快捷,自动化程度高
- 逐步法:适合处理适中的数据量,可以自定义参数
- 分步法:适合处理较大的数据量(5000个以上基因),需要分不同的block划分模块,自定义参数
下面先逐步说一下构建表达网络的步骤。
2.0 参数设置
#与第一步设置一样
getwd();
workingDir = "."; #必须是文件的工作路径
setwd(workingDir);
library(WGCNA)
options(stringsAsFactors = FALSE);
enableWGCNAThreads() #开启多线程
载入第一步保存的数据
lnames = load(file = "FemaleLiver-01-dataInput.RData");
lnames2.1 构建自动化网络和检测模块
选择软阈值
powers = c(c(1:10), seq(from = 12, to=20, by=2))
sft = pickSoftThreshold(datExpr, powerVector = powers, verbose = 5
sizeGrWindow(9, 5)
par(mfrow = c(1,2));
cex1 = 0.9;无标度拓扑拟合指数
plot(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2],
xlab="Soft Threshold (power)",ylab="Scale Free Topology Model Fit,signed R^2",type="n",
main = paste("Scale independence"));
text(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2],
labels=powers,cex=cex1,col="red");
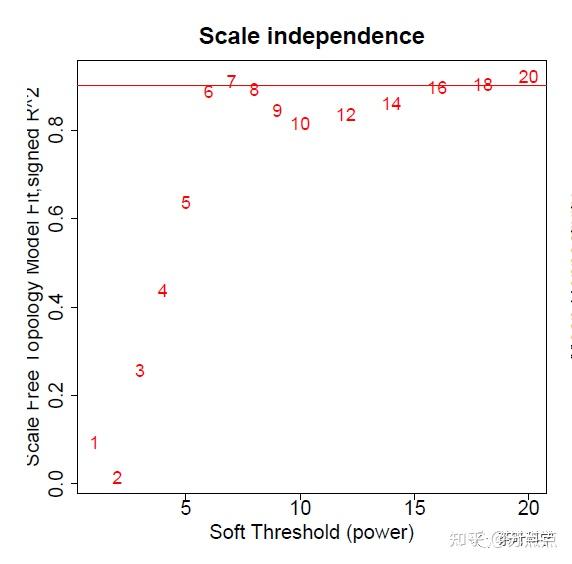
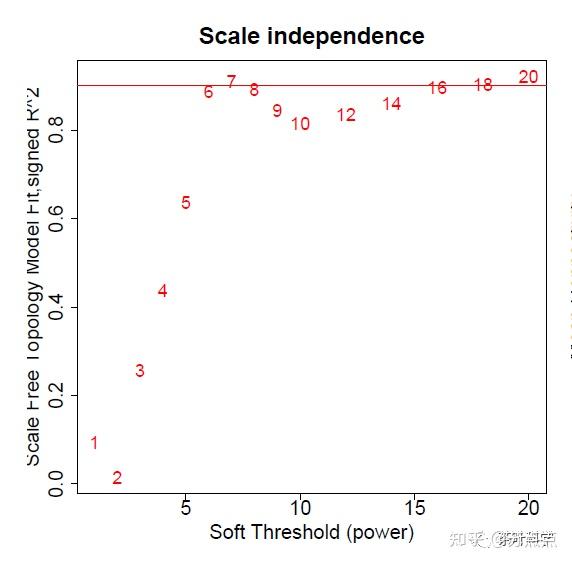
abline(h=0.90,col="red") #查看位于0.9以上的点,可以改变高度值一般来说,无标度拓扑拟合指数这个图是用来选择软阈值的一个根据。例如下图是1到20,有些教程会写到1到30。 我们一般选择在0.9以上的,第一个达到0.9以上数值。下图的6是第一个达到0.9的数值,可以考虑6作为软阈值。
如果在0.9以上就没有数值了,我们就不要降低标准,但是最低不能小于0.8。
平均连接度
plot(sft$fitIndices[,1], sft$fitIndices[,5],
xlab="Soft Threshold (power)",ylab="Mean Connectivity", type="n",
main = paste("Mean connectivity"))
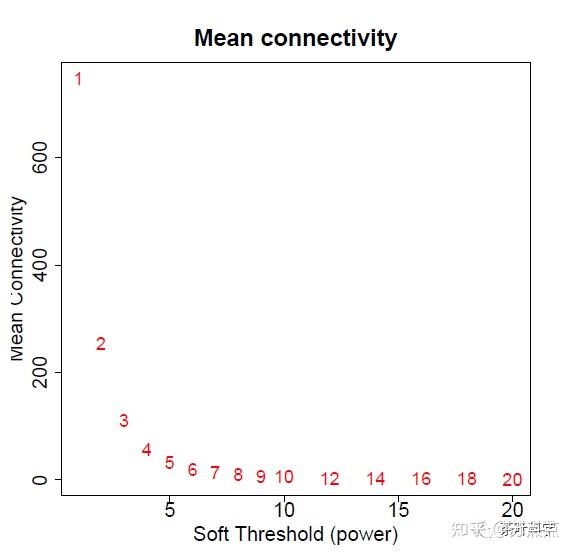
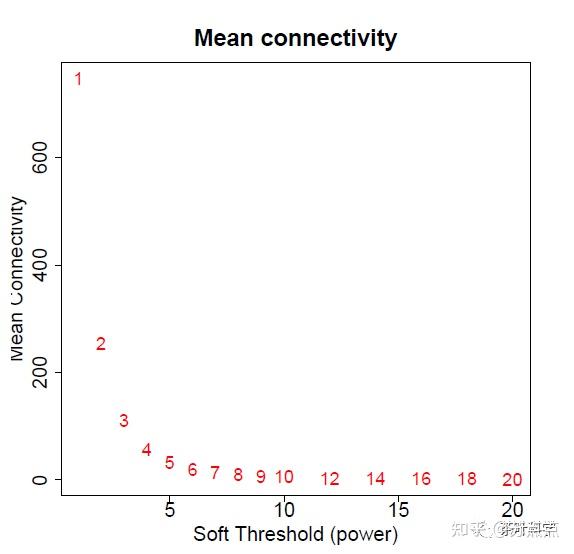
text(sft$fitIndices[,1], sft$fitIndices[,5], labels=powers, cex=cex1,col="red")从下图可以看出,数值为6的时候,已经开始持平,则软阈值为6时,网络的连通性好。
运行下面的代码,如果有合适的软阈值,系统会自动推荐给你。
sft$powerEstimate
#结果为6
如果显示的结果为 NA,则表明系统无法给出合适的软阈值,这时候就需要自己挑选软阈值。
手动挑选软阈值的大致规则如上面所述。关于如何手动挑选软阈值的问题我会放到WGCNA分析进阶版常见问题整理这篇推文里。
如无合适软阈值时,可以按以下条件选择:
if (is.na(power)){
power = ifelse(nSamples<20, ifelse(type == "unsigned", 9, 18),
ifelse(nSamples<30, ifelse(type == "unsigned", 8, 16),
ifelse(nSamples<40, ifelse(type == "unsigned", 7, 14),
ifelse(type == "unsigned", 6, 12))
}
由于篇幅问题,下面只说较为常用的一步法构建网络的步骤。如需要学习这三种不同的方法,可以看 WGCNA分析进阶版常见问题整理 这篇推文。
2.2 一步法构建网络和模块检测
net = blockwiseModules(datExpr, power = 6,
TOMType = "unsigned", minModuleSize = 30,
reassignThreshold = 0, mergeCutHeight = 0.25,
numericLabels = TRUE, pamRespectsDendro = FALSE,
saveTOMs = TRUE,
saveTOMFileBase = "femaleMouseTOM",
verbose = 3)
# power = 6是刚才选择的软阈值
#minModuleSize:模块中最少的基因数
#mergeCutHeight :模块合并阈值,阈值越大,模块越少(重要)
#saveTOMs = TRUE,saveTOMFileBase = "femaleMouseTOM"保存TOM矩阵,名字为"femaleMouseTOM"
#net$colors 包含模块分配,net$MEs 包含模块的模块特征基因。这个例子中保留较多的默认参数,可以在environment包中阅读帮助文档,调整网络结构和模块检测参数以使结果更符合生物学意义。
注意:maxBlockSize的参数默认的模块处理最大基因数为位5000,如果大于5000,这个函数会将数据集拆分为几块,这会破坏下面的一些绘图代码,即执行代码会导致错误。希望分析更大数据集的读者需要执行以下操作之一:4GB运行内存可以处理8000~10000个,16GB最多可处理20000个,32GB最多可以处理30000个。如果要分析较大的数据集,需要用第三种构建网络的方法,即分模块,逐块分析。
查看划分的模块数和每个模块里面包含的基因个数
table(net$colors)
# 0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
# 99 609 460 409 316 312 221 211 157 123 106 100 94 91 77 76 58 47 34
以上结果表示一共可以分为18个模块,第二行是每个模块对应的基因数,有多到少。
从模块1开始,基因数逐渐减少。模块0是无法识别的基因数。模块标识的层次聚类树状图,可以使用以下代码将树状图与颜色分配一起显示:
sizeGrWindow(12, 9)
mergedColors = labels2colors(net$colors)
plotDendroAndColors(net$dendrograms[[1]], mergedColors[net$blockGenes[[1]]],
"Module colors",
dendroLabels = FALSE, hang = 0.03,
addGuide = TRUE, guideHang = 0.05)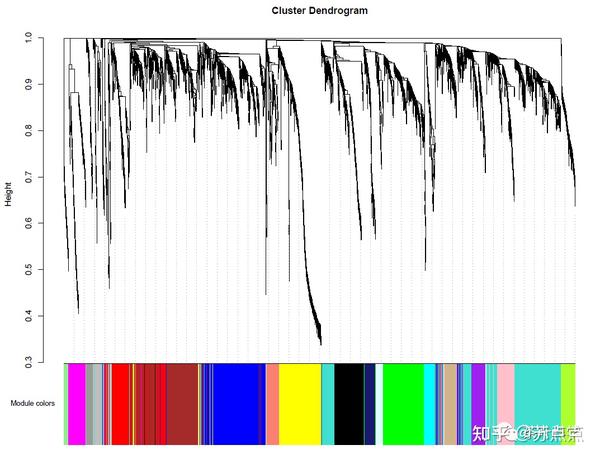
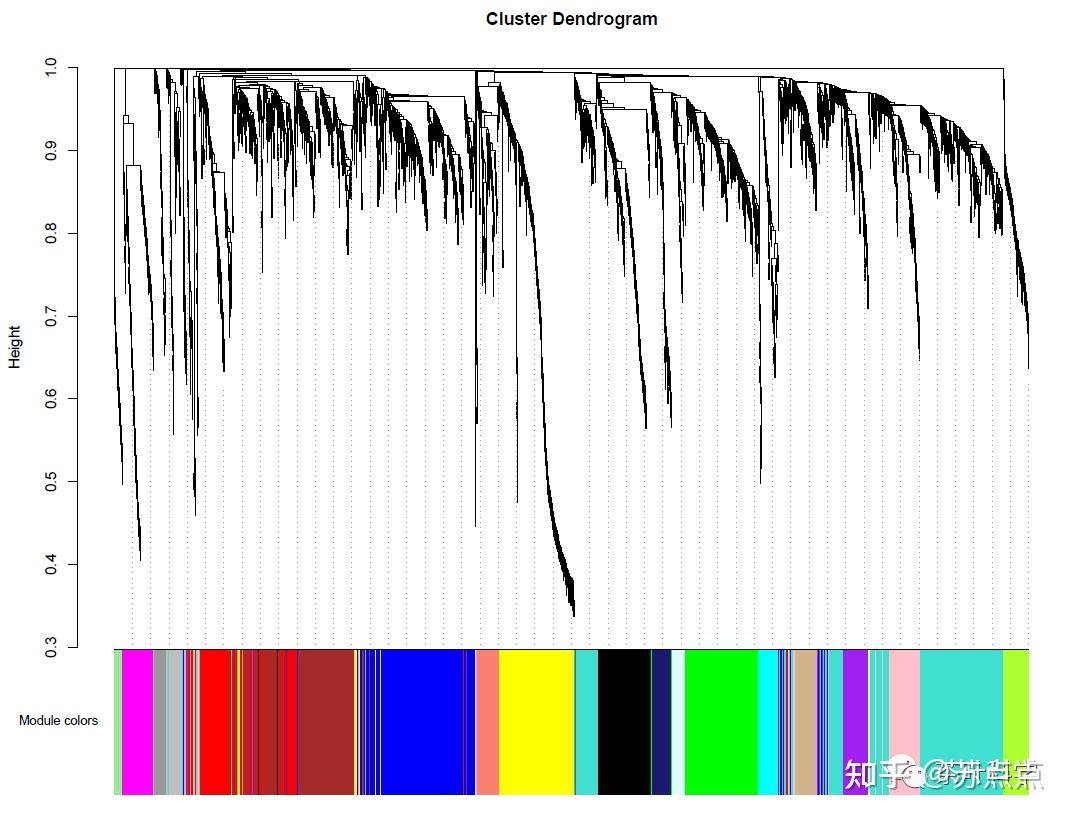
如果需要修改树、模块成员、和模块合并标准,该包的recutBlockwiseTrees函数可以对结果进行修改,而无需重复计算网络和树状图。(推荐用第二种方法 分步法 实现)
保存分配模块和模块包含的基因信息。
moduleLabels = net$colors
moduleColors = labels2colors(net$colors)
MEs = net$MEs;
geneTree = net$dendrograms[[1]];
save(MEs, moduleLabels, moduleColors, geneTree,
file = "FemaleLiver-02-networkConstruction-auto.RData")3. 模块与表型数据关联并识别重要基因
3.0 参数设置与载入之前的分析结果
getwd();
workingDir = ".";
library(WGCNA);
options(stringsAsFactors = FALSE);
lnames = load(file = "FemaleLiver-01-dataInput.RData");
lnames;
lnames = load(file = "FemaleLiver-02-networkConstruction-auto.RData");
lnames这里使用的是一步法自动构建网络的分析结果,也可以用分步法、逐步法的分析结果。
3.1 模块-表型数据关联
在这个分析中,我们将识别与表型数据显著相关的模块。由于我们已经有每个模块的eigengene,只需要将eigengene与外部数据相关联,寻找重要的关联。
nGenes = ncol(datExpr);
nSamples = nrow(datExpr);
# 重新计算带有颜色标签的模块
MEs0 = moduleEigengenes(datExpr, moduleColors)$eigengenes
MEs = orderMEs(MEs0)
moduleTraitCor = cor(MEs, datTraits, use = "p");
moduleTraitPvalue = corPvalueStudent(moduleTraitCor, nSamples);
# 通过相关值对每个关联进行颜色编码
sizeGrWindow(10,6)
# 展示模块与表型数据的相关系数和 P值
textMatrix = paste(signif(moduleTraitCor, 2), "\n(",
signif(moduleTraitPvalue, 1), ")", sep = "");
dim(textMatrix) = dim(moduleTraitCor)
par(mar = c(6, 8.5, 3, 3));
# 用热图的形式展示相关系数
labeledHeatmap(Matrix = moduleTraitCor,
xLabels = names(datTraits),
yLabels = names(MEs),
ySymbols = names(MEs),
colorLabels = FALSE,
colors = greenWhiteRed(50),
textMatrix = textMatrix,
setStdMargins = FALSE,
cex.text = 0.5,
zlim = c(-1,1),
main = paste("Module-trait relationships"))
#colors = greenWhiteRed(50)不适用于红绿色盲患者,建议用 blueWhiteRed代替.
#该分析确定了几个重要的模块-特征关联。我们将体重作为感兴趣的特征来研究。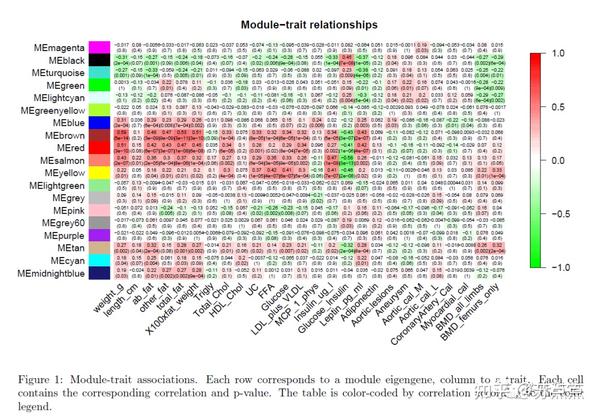
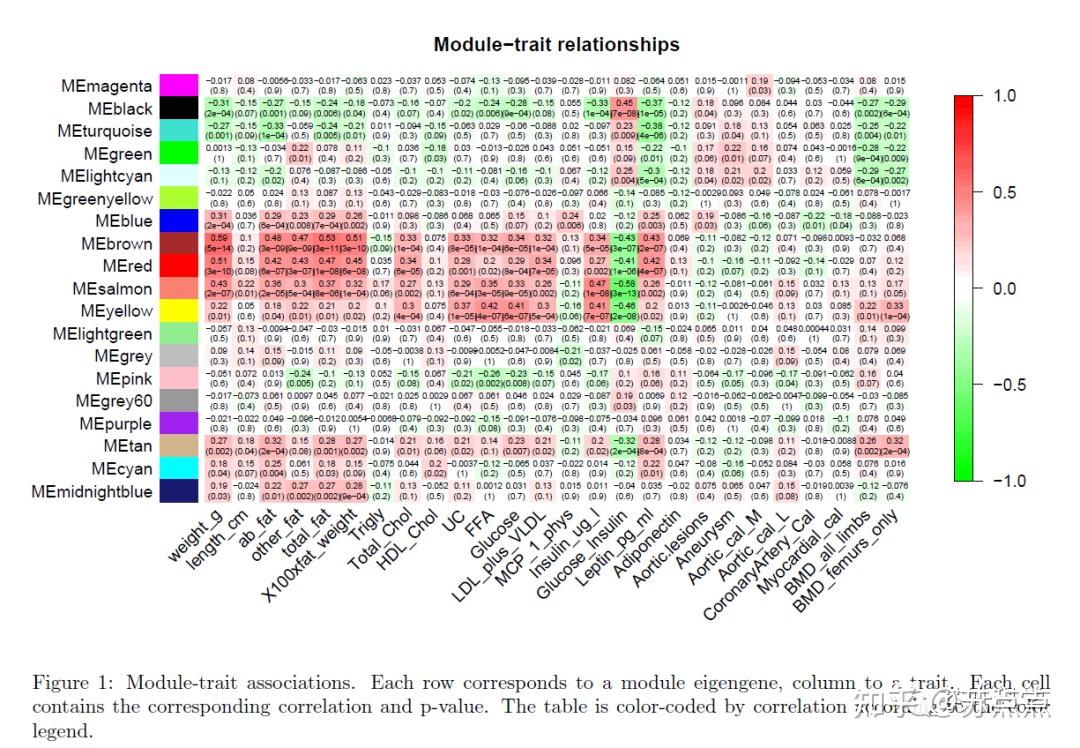
从图片可以看到颜色越红的模块表示与表型性状与该模块的基因高度正相关,而言是越绿表示高度负相关。 可以看到棕色模块与体重的相关性非常高,所以下面会接着探讨这个模块中的基因与体重的关系。
3.2 基因与表型数据的关系、重要模块:基因显著性和模块成员
我们将用基因的显著性GS定义为基因与性状的相关性(绝对值),以定量单个基因与我们感兴趣的性状的关联。对于每个模块,我们将用模块成员MM的定量测定定义为模块eigengene和基因表达特征的相关性。这样能够量化矩阵上所有基因和每个模块的相似性。
weight = as.data.frame(datTraits$weight_g);
names(weight) = "weight";
modNames = substring(names(MEs), 3)
geneModuleMembership = as.data.frame(cor(datExpr, MEs, use = "p"));
MMPvalue = as.data.frame(corPvalueStudent(as.matrix(geneModuleMembership), nSamples));
names(geneModuleMembership) = paste("MM", modNames, sep="");
names(MMPvalue) = paste("p.MM", modNames, sep="");
geneTraitSignificance = as.data.frame(cor(datExpr, weight, use = "p"));#和体重性状的关联
GSPvalue = as.data.frame(corPvalueStudent(as.matrix(geneTraitSignificance), nSamples));
names(geneTraitSignificance) = paste("GS.", names(weight), sep="");
names(GSPvalue) = paste("p.GS.", names(weight), sep="");3.3 模块内分析:鉴定具有高GS和高MM的基因
使用GS和MM测量,可以识别与体重高度相关的基因,以及感兴趣的模块中的高度相关的成员。这个例子中, 体重与棕色模块 的关联度较高,因此我们在棕色模块中绘制基因显著性和模块成员关系的散点图。 注:
GS:所有基因表达谱与这个模块的eigengene的相关性(cor)。 每一个值代表这个基因与模块之间的关系。如果这个值的绝对值接近0,那么这个基因就不是这个模块中的一部分,如果这个值的绝对值接近1,那么这个基因就与这个模块高度相关。
MM:基因和表型性状比如体重之间的相关性的绝对值。 为了将表型特征信息与共表达网络联合起来,比如体重与哪个模块高度相关。每一个基因的表达值与表型性状之间的相关性的绝对值。0表示这个基因与这个性状不相关,1表示高度相关。如果一个模块中的基因都有这个性状高度相关,那么这个模块也就与这个性状高度相关。
运行以下代码可视化GS和MM
module = "brown"
column = match(module, modNames);
moduleGenes = moduleColors==module;
sizeGrWindow(7, 7);
par(mfrow = c(1,1));
verboseScatterplot(abs(geneModuleMembership[moduleGenes, column]),
abs(geneTraitSignificance[moduleGenes, 1]),
xlab = paste("Module Membership in", module, "module"),
ylab = "Gene significance for body weight",
main = paste("Module membership vs. gene significance\n"),
cex.main = 1.2, cex.lab = 1.2, cex.axis = 1.2, col = module)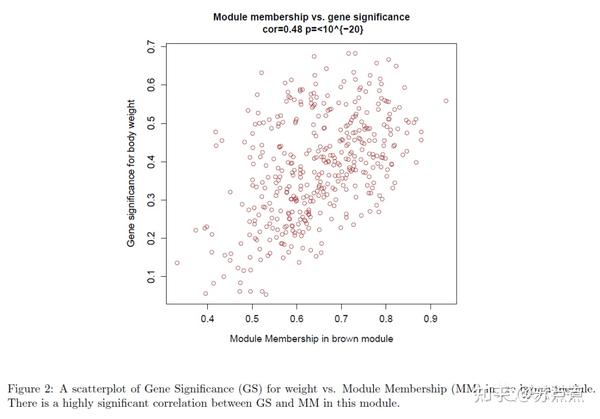
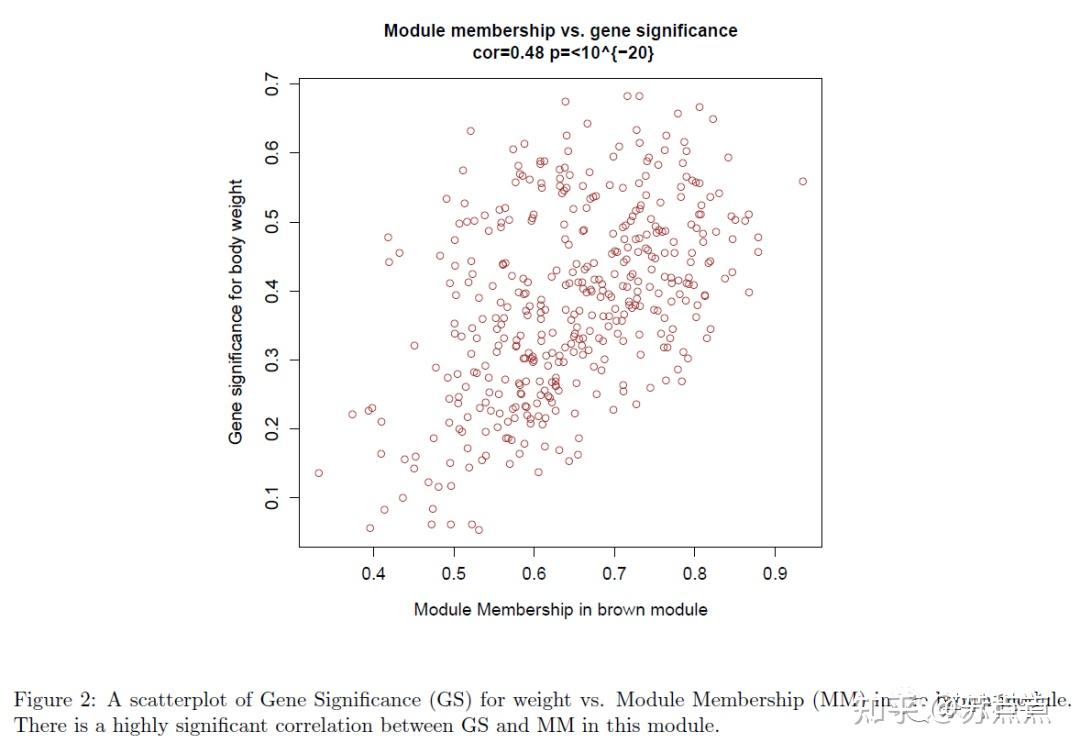
MM-GS图的每一个点:
图中的每一个点代表一个基因,横坐标值表示基因与模块的相关性,纵坐标值表示基因与表型性状的相关性,这里可以看出与性状高度显著相关的基因往往是与这个性状显著相关的模块中的重要元素。
3.4 输出网络分析结果
我们在找到了与我们感兴趣的表征高度相关的模块,并通过MM测量确定了核心的参与者(核心基因hubgene),我们现在需要将这些数据合并起来,并输出为结果文件。
names(datExpr)#会返回所有在分析中的基因ID
names(datExpr)[moduleColors=="brown"]#返回属于棕色模块的基因ID
annot = read.csv(file = "GeneAnnotation.csv");
dim(annot)
names(annot)
probes = names(datExpr) # 匹配信息
probes2annot = match(probes, annot$substanceBXH);
sum(is.na(probes2annot)) # 检测是否有没有匹配上的ID号,正常来说为0,即全匹配上了。
输出必要的信息:
geneInfo0 = data.frame(substanceBXH = probes,
geneSymbol = annot$gene_symbol[probes2annot],
LocusLinkID = annot$LocusLinkID[probes2annot],
moduleColor = moduleColors,
geneTraitSignificance,
GSPvalue);
按照与体重的显著水平将模块进行排序:
modOrder = order(-abs(cor(MEs, weight, use = "p")));
添加模块成员的信息:
for (mod in 1:ncol(geneModuleMembership))
oldNames = names(geneInfo0)
geneInfo0 = data.frame(geneInfo0, geneModuleMembership[, modOrder[mod]],
MMPvalue[, modOrder[mod]]);
names(geneInfo0) = c(oldNames, paste("MM.", modNames[modOrder[mod]], sep=""),
paste("p.MM.", modNames[modOrder[mod]], sep=""))
geneOrder = order(geneInfo0$moduleColor, -abs(geneInfo0$GS.weight)); # 排序
geneInfo = geneInfo0[geneOrder, ]
输出为CSV格式,可用fix(geneInfo)在R中查看:
write.csv(geneInfo, file = "geneInfo.csv")4. 网络交互分析(GO注释等)
这步其实是可选步骤,主要是将重要的基因进行功能注释,例如GO注释。
4.0 参数设置与载入数据
getwd();
workingDir = ".";
setwd(workingDir);
library(WGCNA)
options(stringsAsFactors = FALSE);
lnames = load(file = "FemaleLiver-01-dataInput.RData");
lnames
lnames = load(file = "FemaleLiver-02-networkConstruction-auto.RData");
lnames我们之前分析了与体重weight高度相关的模块,为了有助于阐释在生物学上的意义,我们希望知道这些模块里的基因的基因本体,它们是否显著富集在某些功能类别中。
4.1 输出基因列表供在线软件服务使用
其中最简单的一种选择是导出基因识标符列表,该列表可以在几个常用的基因本体David和功能富集分析AmiGo中输入使用。例如,将brown棕色模块的LocusLinkID(entrez)标识符代码写到一个文件中:
annot = read.csv(file = "GeneAnnotation.csv");
probes = names(datExpr);
probes2annot = match(probes, annot$substanceBXH);
allLLIDs = annot$LocusLinkID[probes2annot];
intModules = c("brown", "red", "salmon")
for (module in intModules)
# Select module probes
modGenes = (moduleColors==module);
# Get their entrez ID codes
modLLIDs = allLLIDs[modGenes];
# Write them into a file
fileName = paste("LocusLinkIDs-", module, ".txt", sep="");
write.table(as.data.frame(modLLIDs), file = fileName,
row.names = FALSE, col.names = FALSE)
}4.2 直接用R进行GO富集分析
WGCNA包包含了执行GO富集分析功能,但是要运行GO富集,需要安Biconductororg包的GO.db、AnnotationDBI和适当的生物体特定注释包。
注: 特定生物体的包以org.Xx.eg.db的形式命名。Xx代表生物体代码:Mm是小鼠、Hs是人类。酵母的注释包比较特别,用的是org.Sc.sgd.db。请访问 http://www. bioconductor.org 的 Bioconductor 主页下载并安装所需的软件包。
这个例子中研究的是小鼠的基因表达,所以要用到org.Mm.eg.db包。 调用GO富集分析函数GOenrichmentAnalysis非常简单,该函数采用模块标签向量,以及给出标签的基因的Entrez(Locus Link)编码。
if (!require("BiocManager", quiet = TRUE))
+ install.packages("BiocManager")
GOenr = GOenrichmentAnalysis(moduleColors, allLLIDs, organism = "mouse", nBestP = 10)
tab = GOenr$bestPTerms[[4]]$enrichment
以上结果是一个富集列表,包含每个模块颜色中10个最佳的条目。可以通过以下方式访问表中列的名称:
names(tab)
输出为*.CSV格式可用Excel打开
write.table(tab, file = "GOEnrichmentTable.csv", sep = ",", quote = TRUE, row.names = FALSE)
也可以直接删减一些内容,使其在R中快速显示出来:
keepCols = c(1, 2, 5, 6, 7, 12, 13);
screenTab = tab[, keepCols];
numCols = c(3, 4);
screenTab[, numCols] = signif(apply(screenTab[, numCols], 2, as.numeric), 2) #将数字保留两位小数
将术语名称截断为最多 40 个字符:
screenTab[, 7] = substring(screenTab[, 7], 1, 40)
colnames(screenTab) = c("module", "size", "p-val", "Bonf", "nInTerm", "ont", "term name");
rownames(screenTab) = NULL;
options(width=95)
screenTab5. 网络可视化
5.0 参数设置
getwd();
workingDir = ".";
setwd(workingDir);
library(WGCNA)
options(stringsAsFactors = FALSE);
lnames = load(file = "FemaleLiver-01-dataInput.RData");
lnames
lnames = load(file = "FemaleLiver-02-networkConstruction-auto.RData");
lnames
nGenes = ncol(datExpr)
nSamples = nrow(datExpr)5.1 可视化基因网络
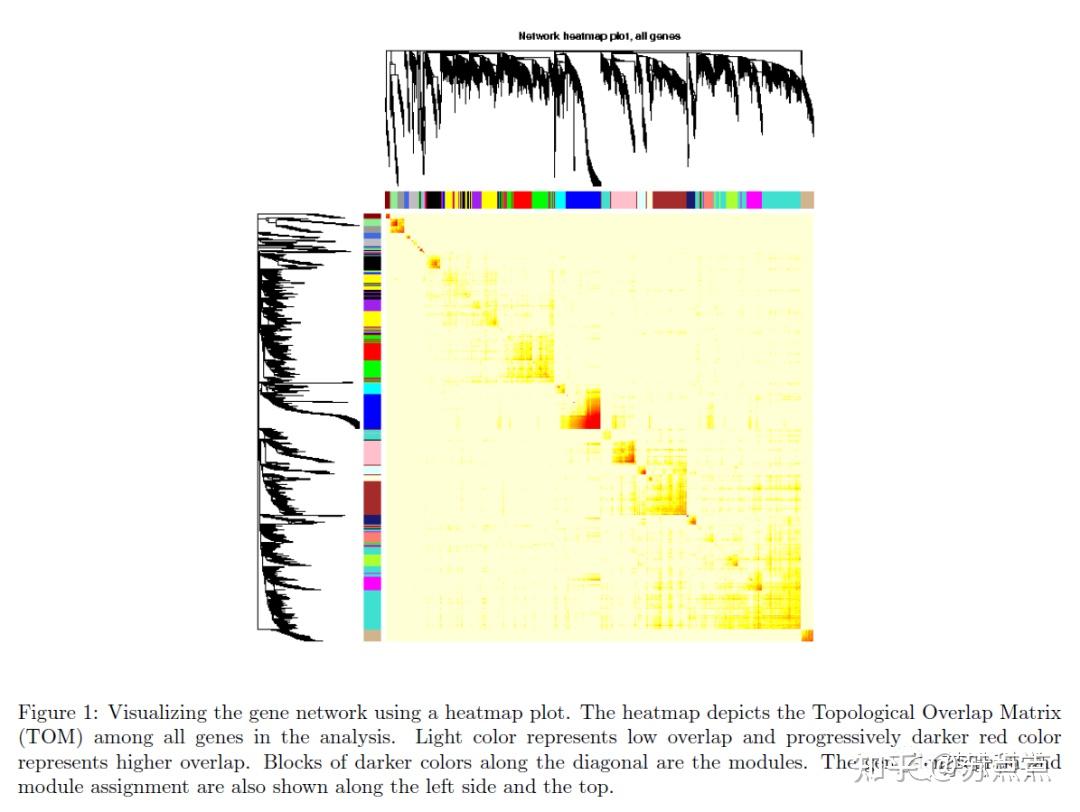
可视化加权网络的方法之一是制作热图。热图的每行每列代表一个基因,浅色代表低邻接(重叠);深色代表高邻接(重叠)。 以下代码是将一步法和逐步法的基础上绘制的热图,不适用于逐块分析法 ,如需要展示逐步法,需要修改代码将每块block进行可视化。
#计算TOM矩阵
dissTOM = 1-TOMsimilarityFromExpr(datExpr, power = 6);
plotTOM = dissTOM^7;
diag(plotTOM) = NA;
sizeGrWindow(9,9)
TOMplot(plotTOM, geneTree, moduleColors, main = "Network heatmap plot, all genes")生成的热图可能需要大量的时间。可以限制基因的数量来加快绘图。但是基因子集的树状图看起来与所有基因的树状图不同, 下面随机选取400个基因进行绘图:
nSelect = 400
set.seed(10);
select = sample(nGenes, size = nSelect);
selectTOM = dissTOM[select, select];
selectTree = hclust(as.dist(selectTOM), method = "average")
selectColors = moduleColors[select];
sizeGrWindow(9,9)
plotDiss = selectTOM^7;
diag(plotDiss) = NA;
TOMplot(plotDiss, selectTree, selectColors, main = "Network heatmap plot, selected genes")
#改变热图的深色背景为白色背景:
library(gplots) # 需要先安装这个包才能加载。
myheatcol = colorpanel(250,'red',"orange",'lemonchiffon')
TOMplot(plotDiss, selectTree, selectColors, main = "Network heatmap plot, selected genes", col=myheatcol)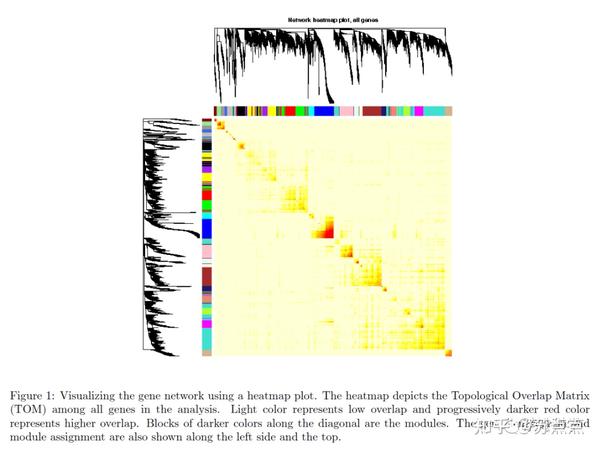
5.2 可视化表征基因网络
研究找到的模块之间的关系,可以使用eigengene表征基因作为代表轮廓,通过特征基因相关性来量化模块的相似性。该包包含一个函数plotEigengeneNetworks,可以生成eigengene网络的摘要图。
# 重新计算模块的eigengenes
MEs = moduleEigengenes(datExpr, moduleColors)$eigengenes
# 提取体重的表型数据
weight = as.data.frame(datTraits$weight_g);
names(weight) = "weight"
# 加入到相应的模块
MET = orderMEs(cbind(MEs, weight))
sizeGrWindow(5,7.5);
par(cex = 0.9)
plotEigengeneNetworks(MET, "", marDendro = c(0,4,1,2), marHeatmap = c(3,4,1,2), cex.lab = 0.8, xLabelsAngle
= 90)以上结果会生成特征模块与体重数据的聚类图和热图。要想拆分聚类图和热图,可以用以下代码实现。
sizeGrWindow(6,6);
par(cex = 1.0)
plotEigengeneNetworks(MET, "Eigengene dendrogram", marDendro = c(0,4,2,0),
plotHeatmaps = FALSE)
par(cex = 1.0)
plotEigengeneNetworks(MET, "Eigengene adjacency heatmap", marHeatmap = c(3,4,2,2),
plotDendrograms = FALSE, xLabelsAngle = 90)
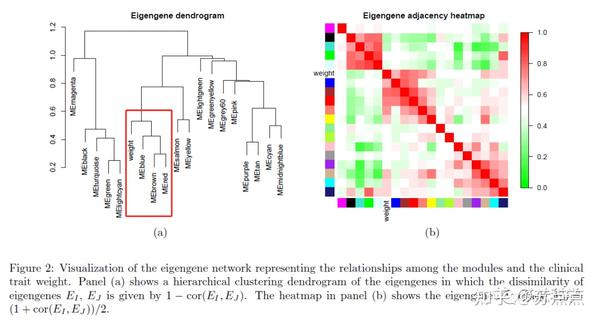
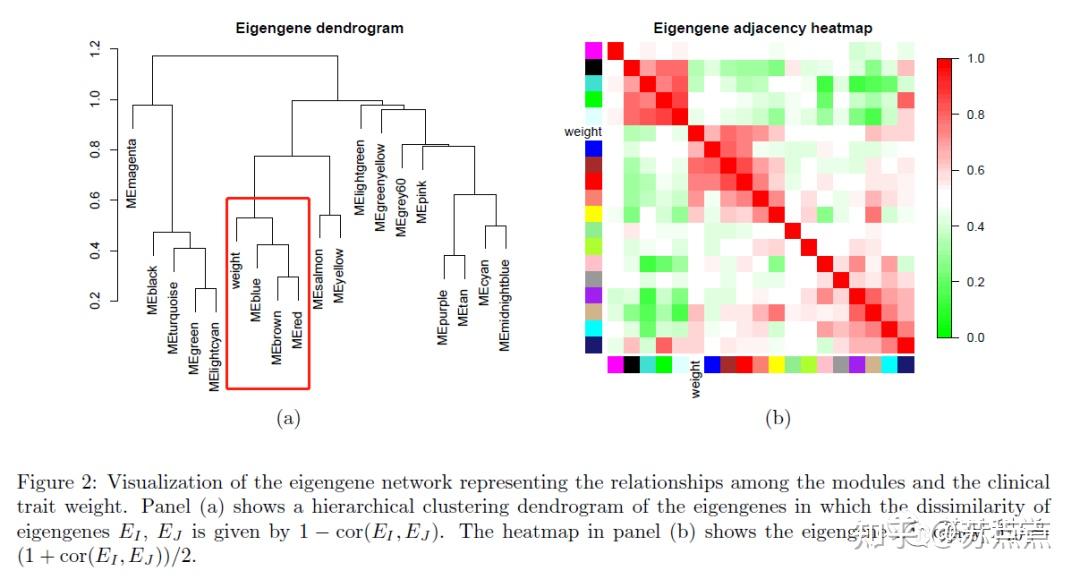
从图中结果可知,体重与模块MEbrown、MEred、MEblue的关系更加密切。
6. 将网络导出到网络可视化软件
第六步是我们最想要的结果,也是每篇文献中最主要的一个图,就是 hub基因的互作关系网络图 。这步会告诉你如何将必要的数据导出,以供其他软件进行绘图,例如VisANT、Cytoscape。
6.0 参数设置与数据导入
getwd();
workingDir = ".";
setwd(workingDir);
library(WGCNA)
options(stringsAsFactors = FALSE);
lnames = load(file = "FemaleLiver-01-dataInput.RData");
lnames
lnames = load(file = "FemaleLiver-02-networkConstruction-auto.RData");
lnames6.1 输出到VisANT软件所需的数据
TOM = TOMsimilarityFromExpr(datExpr, power = 6);
annot = read.csv(file = "GeneAnnotation.csv");
module = "brown";
probes = names(datExpr)
inModule = (moduleColors==module);
modProbes = probes[inModule];
modTOM = TOM[inModule, inModule];
dimnames(modTOM) = list(modProbes, modProbes)
vis = exportNetworkToVisANT(modTOM,
file = paste("VisANTInput-", module, ".txt", sep=""),
weighted = TRUE,
threshold = 0,
probeToGene = data.frame(annot$substanceBXH, annot$gene_symbol) )因为棕色模块相当大,我们可以严格控制输出的hubgene的个数为30个以内在这个模块中。
nTop = 30;nTop = 30;
IMConn = softConnectivity(datExpr[, modProbes]);
top = (rank(-IMConn) <= nTop)
vis = exportNetworkToVisANT(modTOM[top, top],
file = paste("VisANTInput-", module, "-top30.txt", sep=""),
weighted = TRUE,
threshold = 0,
probeToGene = data.frame(annot$substanceBXH, annot$gene_symbol) )以上导出的数据可以用VisANT进行编辑,绘制互作网络。


6.2 输出到Cytoscape
Cytoscape 允许用户输入边缘文件和节点文件,允许用户指定例如连接权重和节点颜色。在这里,我们向 Cytoscape 展示了两个模块(红色和棕色模块)的输出。
TOM = TOMsimilarityFromExpr(datExpr, power = 6);
annot = read.csv(file = "GeneAnnotation.csv");
# 选择棕色和红色的模块
modules = c("brown", "red");
probes = names(datExpr)
inModule = is.finite(match(moduleColors, modules));
modProbes = probes[inModule];
modGenes = annot$gene_symbol[match(modProbes, annot$substanceBXH)];
# 选择相关的 TOM矩阵
modTOM = TOM[inModule, inModule];
dimnames(modTOM) = list(modProbes, modProbes)
# Export the network into edge and node list files Cytoscape can read
cyt = exportNetworkToCytoscape(modTOM,
edgeFile = paste("CytoscapeInput-edges-", paste(modules, collapse="-"), ".txt", sep=""),