


iRegulon:从基因列表到调控网络

其实R里面的包 RcisTarget 也可以完成同样的功能,但基本原理还是在Plos那篇文章里介绍,故而必须精读此文:
PLOS系列发表的文章开头都会有一段非专业术语的描述,这对于入门级选手很有帮助,我很喜欢读:
Gene regulatory networks control developmental, homeostatic, and disease processes by governing precise levels and spatio-temporal patterns of gene expression. Determining their topology can provide mechanistic insight into these processes. Gene regulatory networks consist of interactions between transcription factors and their direct target genes. Each regulatory interaction represents the binding of the transcription factor to a specific DNA binding site near its target gene. Here we present a computational method, called iRegulon, to identify master regulators and direct target genes in a human gene signature, i.e. a set of co-expressed genes.
这里主要讲了几个意思,首先是基因协调网络的重要意义,在生物发育、维持内稳态、疾病发生发展都有重要意义,作者特别提到了基因表达的空间-时间特征。因此,明确基因调控网络的拓扑学有助于对机制的深入探讨。接着是一个重要名词解释,什么叫“基因调控网络”?基因 调控网络就是转录因子跟目标基因的交互作用,这就是所谓的网络生物学了。每一个调控都涉及到了转录因子和目标基因附近特异性基因位点结合。有了这个背景知识后,作者引出了本文的重点在于开发了一个叫iRegulon的软件包,主要用来发现基因标签里目标基因的重要的调控子,这里的基因标签就是一系列共表达基因,可以通过共表达网络来实现,也可以是不同条件下差异表达的基因,总之只要手上有几十到几百个基因列表,iRegulon就会告诉你其中的master调节因子是什么,所谓regulator其实我的理解这里主要指转录因子,是不是还有其它还不清楚。
iRegulon relies on the analysis of the regulatory sequences around each gene in the gene set to detect enriched TF motifs or ChIP-seq peaks, using databases of nearly 10.000 TF motifs and 1000 ChIP-seq data sets or “ tracks ”. Next, it associates enriched motifs and tracks with candidate transcription factors and determines the optimal subset of direct target genes.
那么iRegulon是如何具体工作的呢?我一个小白,怎样能用两句话让我明白?这是相当难度的事情,比开发iRegulon还难。首先是一个富集分析,看一套基因列表是否能富集到motifs或 ChIP-seq peaks,这个motif能够理解,就是 前面学过 的PWM权重嘛,每个TF都对应一个矩阵模型,所有motif不是一个有形的具体的基因序列,而是一个矩阵模型(数学公式),可以输入特定的基因序列,输出一个分数,这个分数代表跟某个TF的结合能。但ChIP-seq peaks就不懂了,这个东西似乎又叫轨迹tracks,既然他里面用or,那暂时理解为跟motif是一个东西,只不过可能不同方法得到的,比如免疫共沉淀测序。这两个都是很大型的数据库。然后富集到的motif就跟已经匹配好的TF相对应起来,同时也确定了最佳的部分基因( subset of direct target genes)。这里为什么是subset呢?基因列表里的基因每个都对应一段上游调控区的片段,这些片段通过PWM模型计算后都可以给出一个分数,因此针对每一个motif,所有基因都可以得到这样的分数,并可以进行排序,因此并不是基因列表里的所有基因都能有很高的分数的。如果在某个TF对应的motif下,基因列表里的大部分基因都获得了很高的分数,比如在所有背景基因(25000个)前3%就把90%的基因列表里的基因给收获(recover)了(现实中90%可能达不到,所以只是一个subset),那么这个motif对应的TF就是这个共表达基因列表的master regulator。
We validate iRegulon on ENCODE data, and use it in combination with RNA-seq and ChIP-seq data to map a p53 downstream network with new predicted co-factors and targets. iRegulon is available as a Cytoscape plugin, supporting human, mouse, and Drosophila genes, and provides access to hundreds of cancer-related TF-target subnetworks or “ regulons ”.
最后聊到验证问题,这个不重要了。iRegulon是Cytoscape的一个插件,能够支持人、鼠、果蝇基因的富集。并且有大量的肿瘤相关的子网络。
简介
Precise regulation of gene expression is imperative for all biological processes. Sequence-specific transcription factors (TFs) bind to their DNA recognition sites within cis -regulatory elements and thereby contribute to the control of the transcriptional initiation rate of their target genes through an interplay with other transcription factors, co-factors, chromatin modifiers, and transcription factories [1] – [3] .
生物体必须要有精确的调控网络才能发挥其作用,序列特异性TF通过结合在顺势作用元件内的DNA识别位点来调节转录,同时还要跟其它很多因子交互,包括其它的转录因子、共同因子等。
The human genome encodes for about 1800 sequence-specific TFs, each of which regulates hundreds of target genes [1] , [4] , [5] . Because TFs play key roles in gene expression, they are often considered the master regulators of cellular processes. Thus, the mapping and characterization of their regulon (all the target genes of a TF) can provide crucial insight into the biological processes they control [6] , [7] . For example, in cancer, ∼40% of the driver mutations affect TFs, and many of the key oncogenes and tumor suppressors, such as p53, MYC, E2F, and NF-κB, are transcription factors [8] .
人类基因组包含了1800个序列特异性的TF,每一个TF调节数百个目标基因。TF就是“大师级调节因子”,而regulon就是某个TF的所有目标基因。举个例子,40%的肿瘤变异基因通过影响TF影响发病机制,一些著名的肿瘤基因如p53, MYC, E2F, 和NF-κB都是转录因子TF。
Identification of the TFs that operate a perturbed gene network, and detecting their target genes, are instrumental steps in uncovering key insights into oncogenic programs, including the discovery of therapeutic targets [9] – [12] . For example, although many target genes have been described for the tumor suppressor p53 [9] , [13] , [14] , several aspects of the gene regulatory network (GRN) downstream of p53 remain unknown. For example, it is still unclear whether p53 also directly represses target genes; whether p53 cooperatively regulates target genes with particular co-factors; and whether different target genes are regulated depending on the cancer type, or depending on the context of p53 activation. The situation is obviously worse for less studied TFs for which often none or only few target genes are known.
p53基因(TF)是研究比较多的基因,但即使如此也仍然有很多未解之迷,比如p53是否直接一直目标基因表达,是否有其他协同因子,在不同肿瘤中目标基因是否不同。其它不太有名的TF就知之更少了。
The targets of a known TF can be identified experimentally with relatively high accuracy through chromatin immunoprecipitation followed by high-throughput sequencing (ChIP-Seq) [15] . However, ChIP-Seq has limitations because it is usually applied to cells in culture rather than to the actual biological sample (e.g., a tumor); and it focuses on a single TF at a time, that has to be chosen a priori . When the TF is not known in advance, or when only gene expression profiling can be performed, regulatory relationships can be uncovered by reverse-engineering a gene regulatory network starting from the expression data.
从已知TF到目标基因的研究,即寻找给定TF的所有目标基因,可以用ChIP-Seq的方法进行。一图了解 ChIP-Seq [1] :
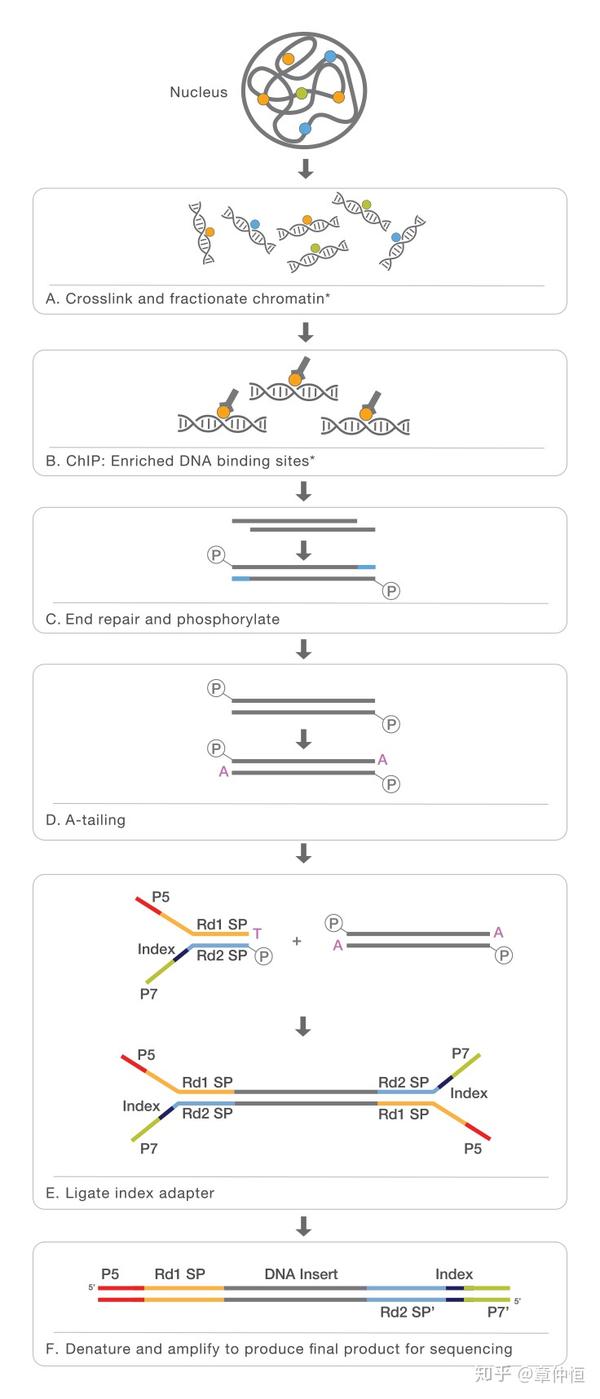

但ChIP-Seq每次只能做一个TF,且需要事先知道TF,这对于只知道基因表达情况下不太适用。
One approach to solve this problem is by exploiting the fact that genes that are co-regulated by the same TF commonly share binding sites for this TF. However, detecting these short and variable TF binding sites (TFBS) within large non-coding regions represents a computational challenge when working with human or mouse genomes. Although a lot of progress has been made over the last decade and many motif discovery methods have been developed and refined (reviewed in [16] – [19] ), motif discovery methods alone are not sufficient to map a gene regulatory network, nor can they be applied to noisy gene sets containing mixtures of targets of multiple TFs. This is true for both motif discovery methods relying on de novo detection and those relying on the enrichment of known position weight matrices (PWM). Additionally, many tools have a motif-oriented output, making it difficult to identify the possible upstream TF. A further limiting factor is that many methods are restricted to using human annotated PWMs (e.g. TRANSFAC [20] , JASPAR [21] or UNIPROBE [22] ), limiting the number of TFs that can be identified as candidate network regulators based on motif enrichment. Therefore, although cis -regulatory sequence analysis has great potential in resolving direct TF-target interactions, it has until today seen limited applications towards gene regulatory network mapping.
一个TF调节的基因通常我结合位点也比较相同,但要从大量的非编码区域检测一小段的绑定区域是个非常大的计算量。过去的十年中motif识别(发现)技术已经有了很大的进步;但光光motif发掘还不够,特别是在绘制完整的调控网络,以及有多个TF参与共同调控网络的时候。这里提到了两个技术:1、依靠重头检测的motif发现技术(是不是就是ChIP-Seq),2、对PWM进行富集的技术。为什么说光有这两个技术还不够呢?技术1一次只能搞一个TF,而PWM富集在多个TF参与调控的情况下可能不行。许多工具输出的结果是motif,还不能最后对应到TF上(这个看了下文后就觉得不是问题)。许多工具都是人纯手工标注的PWM(指标注PWM和那些TF对应),所以TF的量不够大。
Finally, the recent availability of thousands of ChIP-Seq datasets, both from ENCODE [23] , and other resources [24] , yields new opportunities to discover master regulators from co-expressed gene sets [25] , while at the same time pose challenges on how to integrate these data with motif discovery.
根据文中描述,好像有数千个ChIP-Seq,如果理解为每个TF一个ChIP-Seq数据库,那人类总共就1800个TF,那就意味着基本上人类所有的TF都已经建库了。这段话也提示我们,经常出现的著名的ENCODE就是一个ChIP-Seq数据库。这些数据库就可以用来基于共表达基因来发现调控网络。
Here, we aim to tackle some of these challenges by increasing the performance of motif detection to yield high-confidence results, even in noisy gene sets. Motif detection is followed by the annotation of the discovered motifs with associated TFs and direct targets. To this end, we have collected more than nine thousand PWMs from various sources and from different species and link them to candidate binding TFs using a “motif2TF” procedure. This will allow the user to link hitherto anonymous motifs, and motifs of TFs from other species, to candidate human TFs. Furthermore we developed a user-friendly Cytoscape plugin [26] , called iRegulon, allowing the integration of predicted cis -regulatory binding sites directly into a biological network. Finally, we extend and generalize this framework towards combined motif and track discovery on a co-expressed gene set, incorporating more than 1000 ChIP-Seq tracks. The iRegulon Cytoscape plugin is available via the Cytoscape App Store [27] and can be downloaded from
.
本文所开发的算法对于噪声比较高的基因列表也能否很好地发掘基因调控网络。motif富集到之后再进行标注,每个motif对应一个TF。作者从各种数据库中收集了9000多个PWM,这里包含了多个物种,而人是只有1800个TF。这些PWM都对应到了相应的TF。这样用户就能把匿名的motif,其它物种的TF的motif,对应到人的TF上。
The iRegulon framework
The goal of iRegulon is to enable gene regulatory network mapping directly based on motif enrichment in a co-expressed gene set. As motif discovery method we have chosen to elaborate on the recent ranking-and-recovery methods [28] – [32] ( Fig. 1 ). In the ranking step we generate whole-genome rankings of 22284 human RefSeq genes for a library of PWMs where a PWM is a matrix representation of a regulatory motif ( Table 1 ). For each gene, a regulatory search space (500 bp, 10 kb or 20 kb around the Transcription Start Site (TSS), see Materials and Methods ) is scanned for homotypic cis-regulatory modules (CRM) using a Hidden Markov Model [33] ( Fig. S1 ).
iRegulon通过两步完成基因调控网络的绘制。第一步是排序,对22284个人基因进行排序,排序的依据是什么呢?就是PWM库,而一个PWM就是motif的量化表征,是一个模型。对每个基因而言,我们用隐马可夫模型搜寻转录起始点附近(比如500bp)的区域的CRM。
Starting from a library with N PWMs, N ranked lists of genes are generated, each with the most likely genomic targets of a particular motif at the top of the ranking [28] , [29] . Next, orthologous search spaces in ten other vertebrate genomes are determined by UCSC liftover tool [34] and are subsequently scanned with the same PWMs. The rankings for different species are combined by rank aggregation [35] into one final ranking for each PWM in our library. For the PWM libraries we have collected and reformatted most of the available libraries into a “6K collection” ( N = 6383 PWMs) and a “10K collection” ( N = 9713 PWMs) ( Table 1 ). These libraries contain PWMs from different species and also include candidate PWMs for unknown TFs. The results of the ranking step are N human gene rankings stored in an SQLite database. We also generated similar databases using mouse and Drosophila as reference species, in case the input gene set is derived from mouse or fruit fly.
假设有包含N个PWM的库,我们就可以建立N个基因排序,每个基因排序都把最有可能的目标基因排在最前面。然后在10种其它同源物种中重复上述工作,每个PWM的最终排序就通过综合这10个物种的排序获得。这里有两种PWM库,一个是6K库,一个是10K库,主要是包含的PWM数量不同。这些库包含来自不同物种的PWM,以及没与TF对应的PWM。第一个步骤排序完成后,最终结果就是一个SQLite数据库,这个库里包含有N个基因排序。
The recovery step uses as input any set of co-expressed genes ( Fig. 1B ). The enrichment of these genes is determined in each of the N motif-based rankings using the Area Under the cumulative Recovery Curve (AUC), whereby the AUC is computed in the top of the ranking (default set to 3%, see Fig. S2 for validation). The AUC values are normalized into a Normalized Enrichment Score (NES) on which we set a default cutoff of 3.0, corresponding to a False Discovery Rate (FDR) between 3% and 9% ( Fig. S3 and Materials and Methods ). The leading edge of candidate targets is selected as the optimal subset of highly ranked genes compared to the genomic background and compared to the entire motif collection as background ( Fig. 1 B and Materials and Methods ).
第二步是回收,需要共表达基因作为输入信息。就是探讨这些基因在哪些PWM所对应的基因排序中富集,具体可以用累积回收曲线下面积(AUC)来表示。AUC通过计算前3%基因里包含多少比例的共表达基因来表示。这个必须举个例子才能讲清楚。前3%基因就是 22284\times0.03=669 个基因,假设共表达基因有100个,其中80个在前669个基因里面,于是其 AUC = 80 / 100=0.8 ,这就表示,在排序前3%就回收了80%的基因,可以认为该PWM(motif)对应的(TF)就是调节这批共表达基因的重要因子,然而后面还是需要对这个值进行标准化。


进行验证
验证这个软件的功能也很重要,本文也对iRegulon的预测结果进行了实验验证。
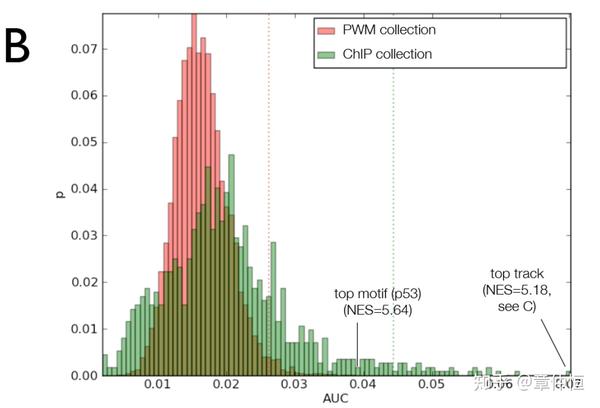
To evaluate the performance of iRegulon, we derived direct target gene sets for 115 sequence-specific TFs from the ENCODE ChIP-Seq data [46] , and for each target set we investigate whether the ChIP'ped TF can be correctly recovered (see Materials and Methods ). Out of 115 tested TFs, iRegulon correctly identifies up to 94 TFs (82.6%) with Normalized Enrichment Scores (NES) above 3 ( Fig. 2A , and Materials and Methods ).
用什么作为验证呢?用115个序列特异性的TF,而这些TF的下游目标基因都是已知的,这些在现成的数据库中都可以获得。验证就是将目标基因集输入iRegulon,返回的TF(根据NES评分对返回的TF进行排序,从而得出top1、top2、top3)与已知的TF进行比较,最完美的状态就是能将iRegulon返回的排第一的TF,与已知给定的TF百分之百对应,而本研究结果显示,iRegulon能将82.6%的TF正确识别,这应该是个不错的数字了。
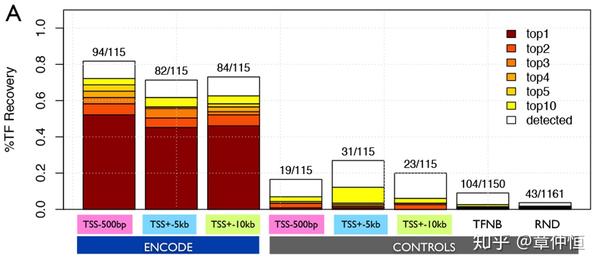

Positive sets consist of the top 200 genes ranked by the maximum signal value of the ChIP-Seq peak in the corresponding search space. Control sets are negatives from ENCODE (genes without a ChIP-Seq peak); TF neighborhoods (TFNB; all TFs within 5 Mb around a query TF); and random signatures (RND). The color (from red to yellow) and order of stacked bars indicate the number of times the queried TF [2] was identified in the 1st rank (top1), 2nd rank (top2), 3rd rank (top3), 4th rank (top4), 5th rank (top5) and 6th to 10th rank (top10). White color indicates the number of detected TFs (motif enrichment ≥3) but with rank >10.
用iRegulon去回收TF。115个TF去做ChIP-Seq,根据信号峰值取前200个基因,对照组里的基因的启动区没有被这115个TF结合(无峰值)。每次用这200个基因输入iRegulon,然后输出每次会对应一批根据NES排序好的PWM(即对应TF)。如果只取其中的第一个TF(top1),则只有50%左右的回收率,top值越大,回收率越大,但可以看到top1已经回收了大部分的TF。而对照组基本无回收,是一个随机回收的状态。


Positives are mixed with negative genes (noise) from 0% to 100% of noise. The lines represent the sensitivity (Sn), Specificity (Sp), and Precision or Positive Predictive Value (PPV) of target gene selection.
观察一个噪声不断增加的连续状态,剂量反应关系在循证医学研究质量评价中有很重要的意义,这里也不例外,随着噪声(阴性基因)的增加,TF回收率不断减少,说明了iRegulon的作用。
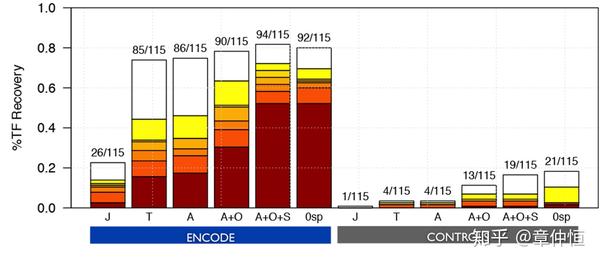

Recovery for ENCODE signatures and their control sets using different motif2TF parameters: 1) Motif collection effect (J, T, A barcharts), 2) Homology effect using a threshold on Identity% for all motifs (A+O barcharts), 3) Motif similarity effect using a threshold on the p-value (A+S barcharts), and combinations (A+O+S). Only Jaspar motifs (J); Only Transfac Pro (T); All motifs from Jaspar and Transfac pro, and others databases (A); All motifs+Orthology (A+O); and All motifs+Orthology+Similarity (A+O+S); blue indicates the analysis done on ENCODE sets and grey indicates on the control sets.
这里探讨了motif与TF对应的问题,看来motif与TF不是一对一的关系,不同的库有不同的motif与TF对应,此处还是不太明白。
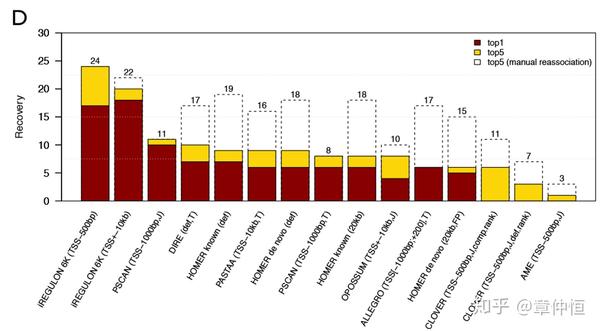

Tool comparison using a benchmark of 30 gene sets constructed as the top 200 target genes based on ChIP peak occurrences in the 20 kb regulatory region for 30 TFs (these TFs were selected from FactorBook having their canonical motif as top enriched in the actual ChIP peaks). The number of times the queried TF was identified in the top1 (red) and top5 (yellow) is recorded. The dashed boxes represent top 5 recoveries if similar motifs are manually re-associated to the query TF. Default parameters were used, but when possible, they were adjusted to use the tss-centered-20 kb regions.
总共30个TF,每个TF选取通过ChIP获得的头200个目标基因,在20kb范围内;这30个TF从FactorBook中选取,特点是其经典motif是经过严格验证的。其它工具似乎跟iRegulon没法相提并论。
Regulons can be discovered from various types of noisy and heterogeneous gene sets
这个题目值得深思,首先Regulons代表了一个 TF调控的所有下游基因,因此我们平时通过共表达网络或差异分析得到的基因列表其实有很多噪声。从这些噪声中识别出信号,就是所谓的Regulons。
In the validation and benchmark analyses above we used gene sets derived from ENCODE ChIP-Seq data as input for iRegulon. In this section, we explore more realistic types of inputs, such as co-expressed genes downstream of a TF perturbation [47] ; genes involved in the same signaling pathway (e.g., KEGG [48] , Reactome [49] or Gene Ontology [50] ); highly connected genes in a biological network (e.g., GeneMania [51] or STRING [52] ); shared targets of a common microRNA. In the first example, we applied iRegulon to a set of 171 genes that are significantly up-regulated under hypoxia [53] . iRegulon yields a top-scoring regulon that contains HIF1A as master regulator, along with 94 predicted direct target genes ( Fig. S5A ). The predicted HIF1A targets are likely functional targets because they overlap much more (41%) with known HIF1A targets [54] than the non predicted targets (15%). More systematically, when applied to 76 co-expressed gene sets obtained after a genetic perturbation of the TF (gene sets from MSigDB [47] ), the perturbed TF is recovered in 38 cases (50%) and as the top ranked master regulator in 18 cases (24%). The lower recall to detect the correct upstream TF compared to ChIP-derived gene sets is expected because not all TF perturbation experiments successfully result in significant gene expression changes of the direct target genes.
这部分验证采用实际的共表达基因来做,有几种获得这些共表达基因的途径,包括通过扰动某个TF、同一个信号通路的基因,生物网络中高度联系在一起的基因,以及由共同的microRNA干扰的基因。
第一个例子是在低氧血症条件下上调的171个基因。这批基因输入iRegulon后,返回了得分最高的一系列基因(Regulon),以及转录因子HIF1A,以及94个直接目标基因:思考一下:这94个基因应该就是通过在3%的背景基因中回收的基因,这时的 AUC=94 /171=0.5497076 ,可能也不是。这里的关键就在于用一个给定的TF是如何去预测目标基因的,通过ChIP-seq?还有一批基因是已知的HIF1A targets。不管怎样,这里的意思就是预测的跟已知的重叠比较多(38/94=41%),而预测的跟非预测的重叠比较少(15%)。
iRegulon can be applied to any kind of gene set to predict upstream regulatory TFs along with significant direct targets, forming TF-target regulons .
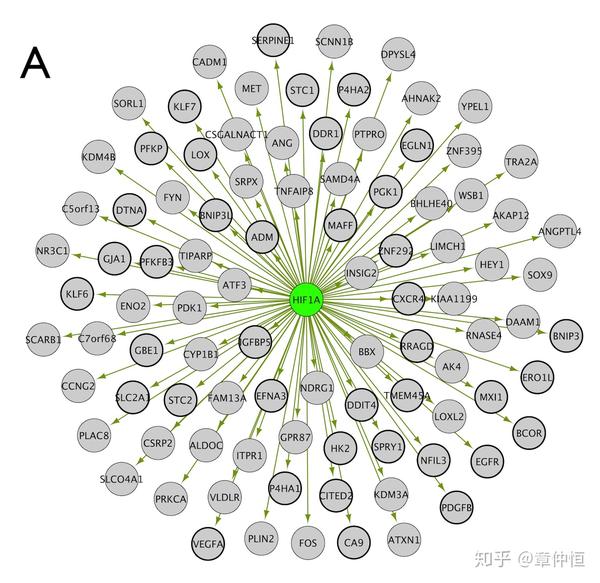

94 HIF1 -\alpha targets identified in 171 genes involved in Hypoxia (11 PWMs, NES = 4.89, rank = 1) (see also Fig. S2 for further details on this iRegulon analysis). Known HIF1A targets [54] are in thick circles.
这里提到的11个PWM是怎么回事?不是说一个TF对应一个PWM吗?或者是理解成:库里的所有PWM都根据回收率计算出一个NES,在前3%的基因里有回收的有11个PWM,而其中NES最高( rank=1 )的motif就是4.89,而 这个motif对应的TF就是HIF1A。目测画粗线的目标基因只有38个,这些已知的可能是各种试验方法获得的基因。而94个是根据生信方法回收到的,这样前面的41%就知道是怎么算出来的了。
第二个例子通过扰动76个TF获得了76套共表达基因,这时TF的回收率偏低,只有50%,而只有18%是将给定的TF作为排名第一的master regulator。原因可能是,并不是所有的TF扰动都能导致下游的目标基因表达发生改变。
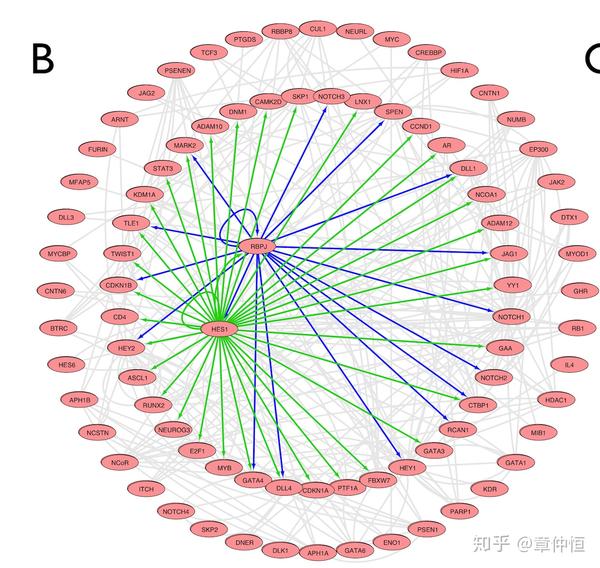
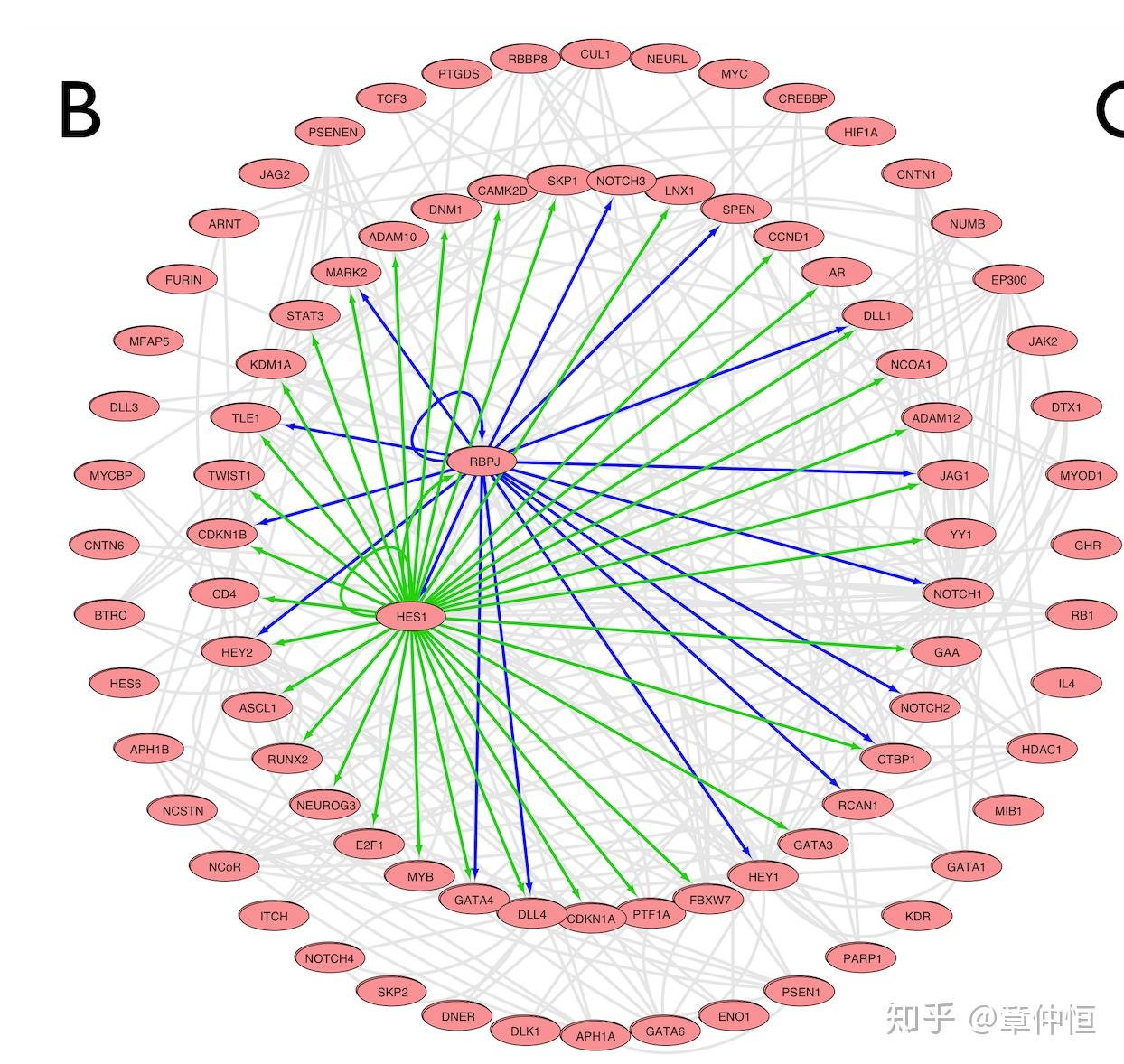
Application to genes from the Notch signaling pathway (Pathway Commons Web Service Client in Cytoscape: NCI/Nature Pathway Interaction Database (ID: notch_pathway)). The imported pathway is composed of 161 molecules and 750 edges. Pathway interactions between genes are in grey and predicted regulatory interactions are in green or blue. We applied iRegulon on all the 87 genes. HES1 (green edges source node) is ranked 1st (NES = 5.099, 5 PWMs) with 35 predicted direct targets. RBPJ (blue edges source node) is ranked 3rd (NES = 4.329, 2 PWMs) with 17 predicted direct targets, including HEY1 , HEY2 , and HES1 . These co-regulators control 47% of the genes if the NOTCH signalling pathway (41/87 genes).
这里是用 iRegulon 对一个已知信号通路里的基因进行分析,这个信号 通路包含 161个分子(可能基因,可能TF),形成了750条边。灰色的边代表基因之间的相互作用,调节作用用绿或蓝色表示,估计中间那两个是TF。把这87个基因输入 iRegulon ,HES1就是排序最高的,回收了35个基因( AUC=35/87=0.40 )。这个 网络由2个TF调控,看来分析一批共表达基因不能只选择NES值最高的,前面几个也许都参与了基因共表达的调节,但选多少个仍然是个疑问。
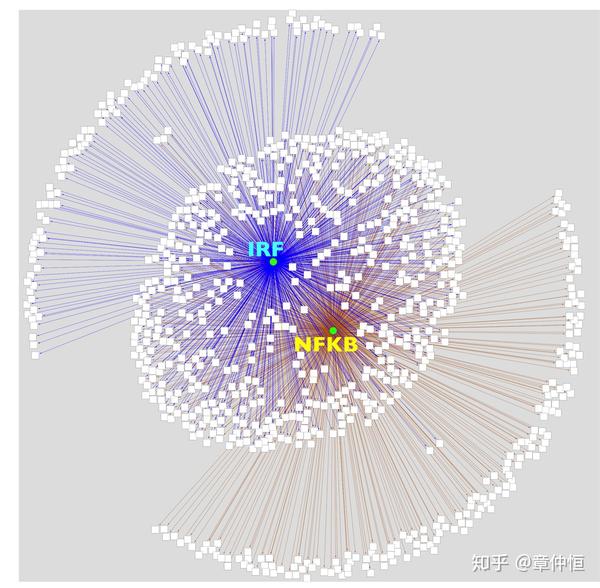
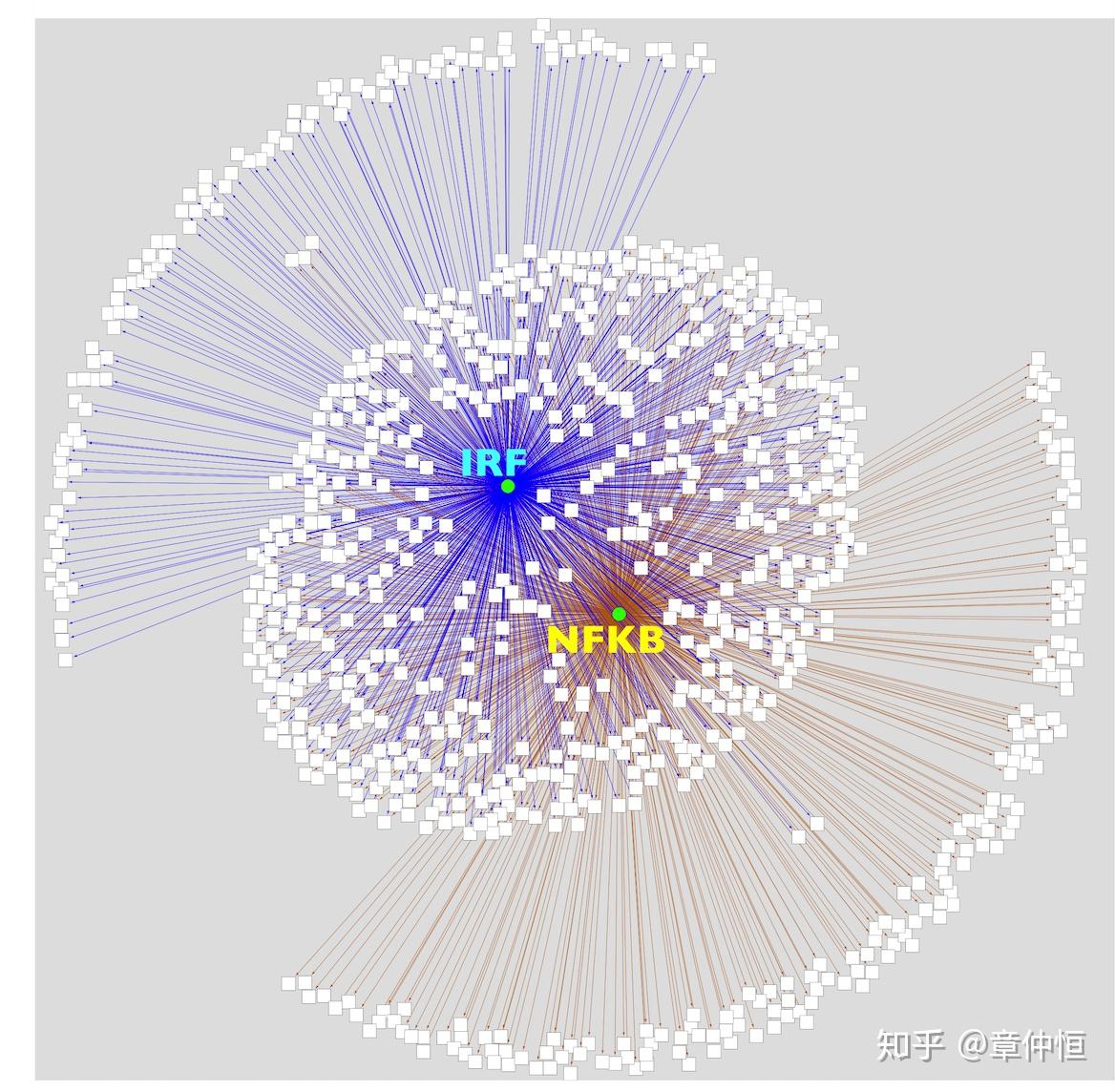
Application to immune response signature. The Immune response gene set is a list of 1923 gene products in Homo sapiens associated to immune response (GO:0006955 and children) was downloaded as a tab delimited file from http://amigo.geneontology.org . Then, this list was converted in a list of 1198 unique gene names (HGNC) and imported in Cytoscape as a network. When applied to these 1198 genes, iRegulon finds the IRF and REL/NFkB regulons, with 806 and 711 direct target genes respectively, indicating that these are indeed that master regulators of the immune response.
这部分做的是免疫反应标签。免疫反应由1923个基因产物参与,这些转化成1198个唯一基因名,用iRegulon去分析发现了IRF和REL/NFkB两个调节子,分别调控了806和711个基因,这几乎覆盖了全部的1198个基因,也证明IRF和REL/NFkB确实是master regulator。
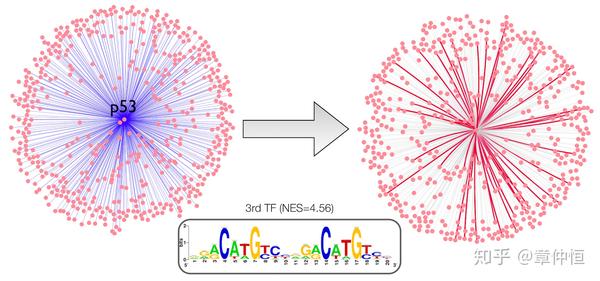
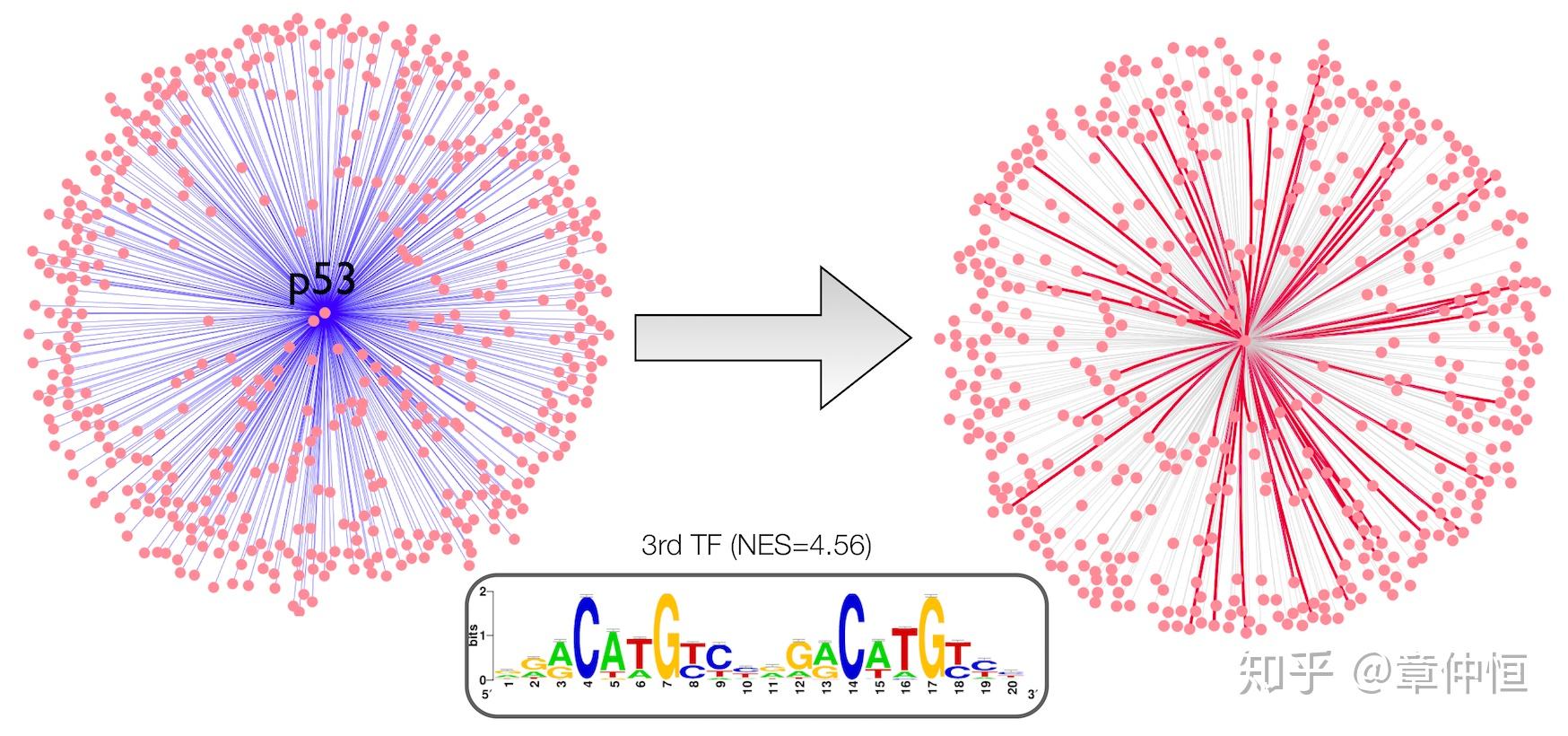
Application to protein-protein interactions from STRING. iRegulon was applied to 500 genes associated with p53 in STRING. The p53 motif was found enriched with an enrichment score of 4.59. Predicted direct interactions are shown in red.
从STRING里提取蛋白交互作用的网络,用iRegulon去分析与p53有关的500个基因,结果发现p53的 NES=4.59,排名第三。预测的在右图显示,预测的基因其实就是前3%回收的基因。存有疑问:这里的蛋白交互似乎跟基因交互没什么差别。
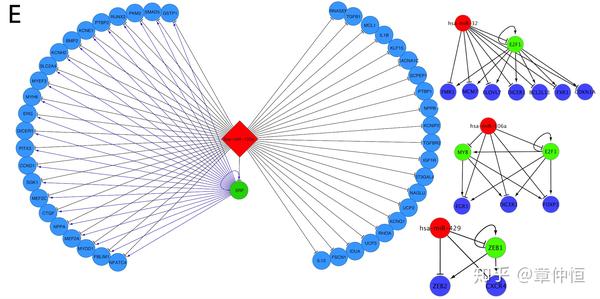

iRegulon analysis has been performed on 159 microRNAs with annotated targetomes. Examples are shown for annotated targets of hsa-miR-133a, has-miR-32, has-miR-429 and has-miR-106a. microRNAs are in red nodes and target nodes are in blue or red (TF). For each microRNA targetome, the enriched TF (found by iRegulon) is represented in green. For example, SRF (green node) was found enriched with a top motif ranked 5th(NES = 4.149) in hsa-miR-133a targetome.
用iRegulon分析了159个标注过的microRNAs,上图显示是其中4个。microRNA作用于基因,通过这些基因寻找到了TF。这些TF应该是事先通过实验或其他手段已知的,这样才有验证iRegulon作用的功能。
While previous methods have thus far been validated and applied to co-expressed gene sets derived from gene expression profiling, here we show that motif discovery with iRegulon can quickly identify master regulons on diverse types of gene sets, as long as a small fraction of the input set is directly co-regulated by the same TF.
这是一个小结。用iRegulon来发现motif能快速找到一系列目标基因的master regulons。输入基因中的一部分是该TF直接调节的。
Mapping a gene regulatory network downstream of p53
We now applied iRegulon to study the gene regulatory network downstream of the p53 tumor suppressor. p53 functions mainly, if not exclusively, as a TF which regulates the expression of hundreds of genes that in turn mediate its biological activities including induction of cell-cycle arrest, senescence and apoptosis [55] , [56] . Although p53 is one of the most-studied transcription factor and hundreds of target genes have already been identified [14] , [55] , many aspects of its downstream network remain unresolved and a more comprehensive understanding of the p53 downstream signaling network is crucial given its importance in oncogenesis.
这部分要做的内容就是用iRegulon去分析p53下游的基因调控网络。为什么选择这个TF呢?因为p53是被广泛研究的,它能调节数以百计的下游基因,但这只是相比较其它还未深入研究的TF而言,p53的许多功能,及其下游网络还是未知的。这段文字只是开个小头,没什么烧脑的干货。
We first determined a p53-dependent gene signature in the MCF-7 human breast cancer cell line by RNA-seq upon stabilization of p53 by the non-genotoxic small molecule Nutlin-3a [57] . This treatment resulted in significant up-regulation of 801 genes and down-regulation of 790 genes. Both up- and down-regulated gene sets were subsequently analyzed with iRegulon ( Fig. 3A ). The top-scoring regulon in the list of up-regulated genes is confirmed as the p53 regulon, with 307 genes predicted to be direct targets ( Fig. 3A and Table S2 ). This indicates that p53 itself is the master regulator of the downstream network and directly controls many up-regulated genes, but not all of them (at least 38%). A Gene Ontology (GO) enrichment analysis of the 307 predicted direct targets identifies p53-related processes and pathways, such as “p53 signaling pathway” (adjusted pvalue = 3.18e-21) or “Apoptosis” (adjusted p-value = 6.76e-07), while the set with the remaining 494 up-regulated genes show no significant GO term enrichment (data not shown).
首先确定p53依赖的基因标签,所谓标签signiture其实就是一系列基因,包括801上调的基因和790下调的基因。这些基因都纳入iRegulon进行分析。其中p53是上调基因的regulon(p53+307个直接目标基因),p53是很重要的调节因子TF,直接控制了 307 / 801=38\% 的基因。富集分析也显示这307个基因是p53信号通路和凋亡。剩余的 801-307=494 个基因没有GO显著性富集。接下去来看看图:
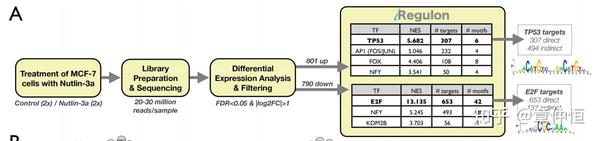

MCF-7 breast cancer cells were treated with Nutlin-3a to stabilize p53, followed by RNA-Seq after 24 h. iRegulon results shows p53 as top regulator in a set of 801 up-regulated genes, represented by 6 significantly enriched motifs, and 307 predicted direct targets. The top regulator in the set of down-regulated genes is E2F, with 653/790 predicted direct targets.
p53是最重要的TF,这个可以理解,但E2F就不太好理解,这个TF可能与p53存在一定的联系?图最右侧的是PWM的logo。表格里的motif个数就不太好理解,不是说一个motif对应一个TF吗?这里信息提示6个motif才对应p53。
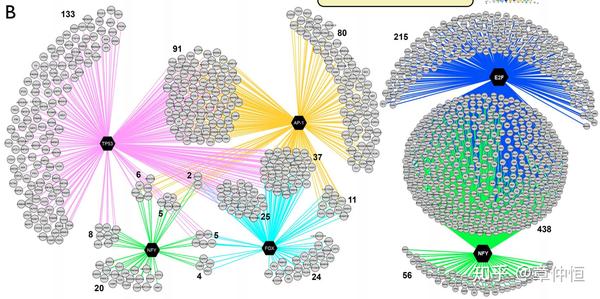

Regulatory network for up-regulated target genes showing the overlap between the p53 regulon and regulons of predicted co-factors (AP-1, NFY, FOX) and regulatory network for down-regulated target genes showing a strong overlap between the predicted E2F and NF-Y regulons. Targets are in grey circle nodes and TF in black hexagon nodes. Regulons for each TF are represented by different edge colours.
上调和下调的基因分开分析,结果发现了不同TF调节的目标基因有很多重叠。
In this particular experimental setup the master regulator, namely p53, was specifically perturbed and thus known a priori . Yet, even under such circumstances there are two important advantages of using a computational regulatory analysis with iRegulon. First, the explicit finding of the p53 motif as top ranked indicates that p53 directly controls a large portion of the up-regulated genes but not all, creating two clearly distinct subsets. Second, we discover potential p53 co-factors and secondary regulons downstream of p53. Particularly, among the 801 genes that are activated downstream of p53, we found three other regulons, one operated by activator protein 1 (AP-1, heterodimer composed of JUN/FOS/FOSL1/FOSL2), another by a Forkhead TF (FOX), and another by NF-Y ( Fig. 3A , Table S3A ). These secondary regulons show extensive overlap with the primary p53 regulon, indicating that these TFs may be important contributors in gene regulation downstream of p53 ( Fig. 3B ). The AP-1 regulon, sharing 136 genes (59% of its regulon) with the p53 regulon might indicate a prevalent co-factorship between the two proteins, something that has been reported before but never on such an extended scale [58] , [59] . In addition, one of the shared p53-AP1 targets is GADD45A , a gene involved in DNA damage repair, that has been shown to be a bona fide target of both p53 and AP-1 [60] . Interestingly, two subcomponents of the AP-1 complex, FOS and FOSL1 , are themselves up-regulated upon p53 stabilization, and are among the predicted direct p53 targets ( Table S4 ). These results, together with the fact that the AP-1 motif was not enriched among the down-regulated genes indicate a positive, synergistic effect of the p53 and AP-1 regulons.
在这些实验中,采用实验去干扰p53,这是事先设定好已知的。用iRegulon分析的好处有:1、p53只控制了一部分的上调基因,这样形成了两个不同的基因亚组;2、有些是p53的共同因子,以及下游的regulon,在上调的801个基因中,除p53外,我们还发现了其它一些regulon,这些次级regulon跟p53有很大的交互重叠。其中AP-1和p53有59%的基因重叠,说明这两个可能是共同因子 ,这在以前的实验中有被证实。除此之外,有个DNA修复基因 GADD45A,是p53和AP-1的共同目标基因。这些结果提示了p53和AP-1具有协同作用。
Nutlin-3a treatment also resulted in 790 significantly down-regulated genes. Interestingly, the analysis of this set with iRegulon does not detect the p53 motif as enriched. It does however identify E2F as master regulator with an astounding 653 (82.7%) predicted direct targets ( Table S3B ). Moreover, three E2F family members, namely E2F1, E2F2, and E2F8 are all strongly and significantly down-regulated upon Nutlin-3a treatment (around 10-fold down with p-value<1.0E-64), indicating the marked involvement of this protein family in the repressive mechanisms of p53. Similarly, iRegulon points towards NF-Y as an important second master regulator of a large number of down-regulated genes (493 genes). Both E2F and NF-Y have been reported as important players for p53-mediated down-regulation of genes [61] , [62] . This may happen through p21 regulated cyclin dependent kinases, resulting in a lack of phosphorylation of NF-Y and Rb which ultimately renders both NF-Y and E2F (through Rb) inactive [63] , [64] . Interestingly, the majority of NF-Y's predicted regulon overlaps with that of E2F, with only a very small number of genes predicted as NF-Y only targets ( Fig. 3B ). The enriched Gene Ontology terms of these overlapping target genes are related to cell-cycle processes, an expected result since both E2F and NF-Y have been established to regulated cell cycle-related genes, often in a cooperative manner [65] – [67] . In contrast to E2F, NF-Y itself is not down-regulated as a gene by p53 activation. However, it is possible that NF-Y is regulated at the protein level rather than at the transcriptional level in response to p53 activation. All together, these findings support the notion of an indirect rather than a direct p53 repressive process largely working through the p53-p21 axis, which affects both E2F and NF-Y [63] , [68] . All together, iRegulon generates marked ideas concerning p53, which are further elaborated upon in the next section.
接下来讲下调的基因,这里首要的TF是E2F,回收了82.7%的下调基因。p53介导下会导致E2F家族(E2F1、E2F2、E2F8)基因表达的下调。p53的抑制作用是通过间接作用实现的。
ChIP-Seq on p53 and E2F confirm their predicted regulons
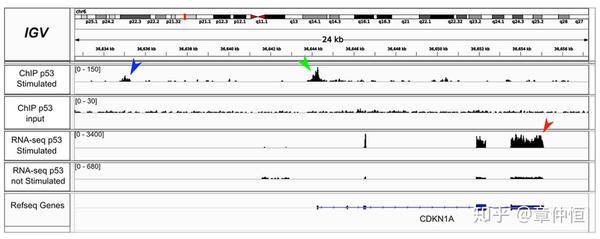
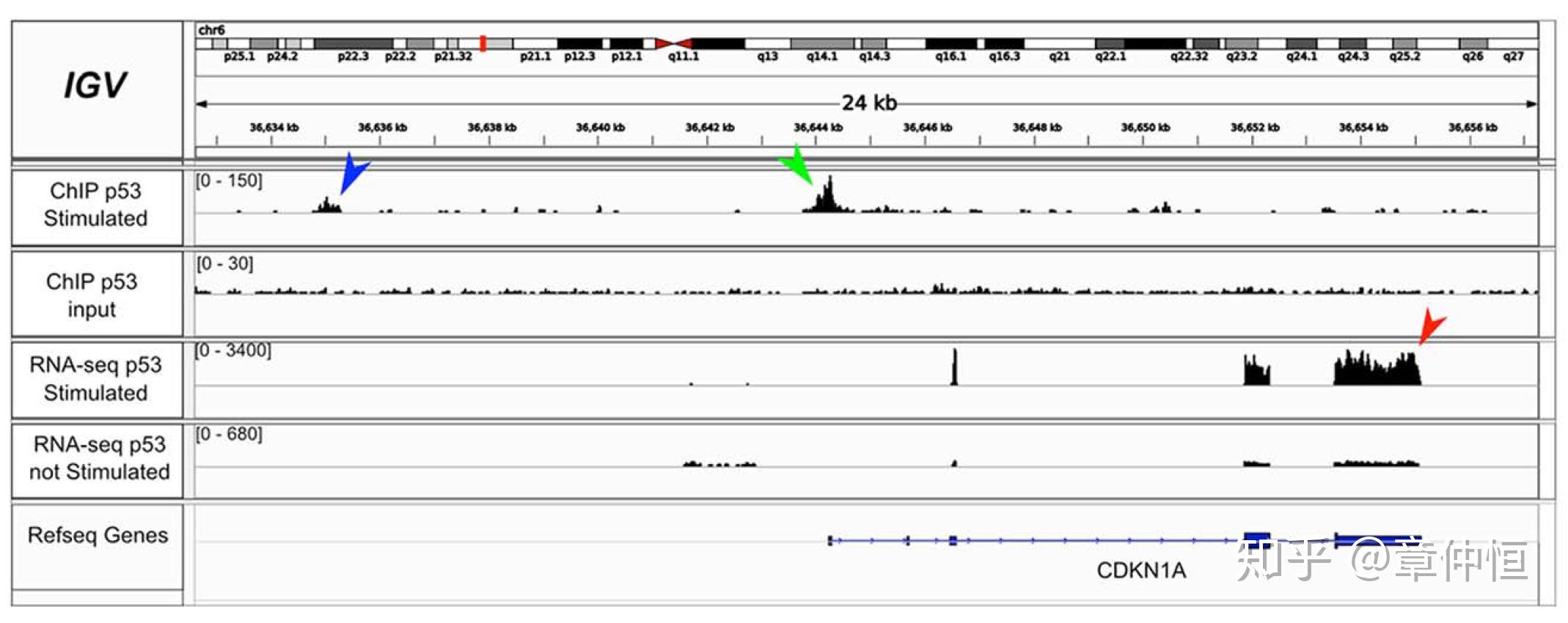
Integrative Genomic Viewer (IGV) [131] screenshot for CDKN1A , a known p53 target gene, showing up-regulation by RNA-seq (red arrowhead) and ChIP peaks in the upstream region (green and blue arrowhead). IGV is free software under GNU Lesser General Public License, version 2.1 (LGPL-2.1).
二代测序结果显示CDKN1A 外显子表达显著增高,在其上游的启动子区用ChiP方法发现信号增强。
To assess whether this ranking yields true p53 targets on top, we curated 223 bona fide p53 targets from the literature and public databases ( Table S5 ), and indeed found these targets to be significantly enriched in the top of this ranking ( Fig. 4B , p-value = 1.40E-24). Within the same ranking, the 307 predicted p53 targets by iRegulon are nearly as significantly enriched in the top as the curated targets (p-value = 2.60E-24), while the 494 remaining up-regulated genes are not significantly correlated with the ChIP peak data (p-value = 0.096). Importantly, this result shows that iRegulon is not only able to identify the master regulator, but is also able to correctly distinguish between direct and indirect targets from a set of co-expressed genes. Only two up-regulated genes with a high ChIP peak, namely PLK3 and DDB2 , were missed by iRegulon. About 100 up-regulated genes have a small ChIP peak but have not been predicted by iRegulon as target genes. These peaks are likely false positive ChIP peaks because they do not show p53 motif enrichment when analyzed separately ( Fig. S6A–C ).
从文献中收集223个确定的p53基因(下图),结果发现这些基因在这个排序(根据ChIP峰值进行排序)中富集。
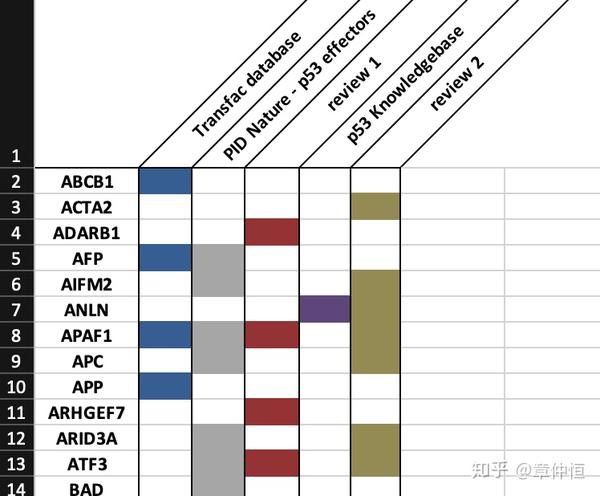

富集结果在下图所示:
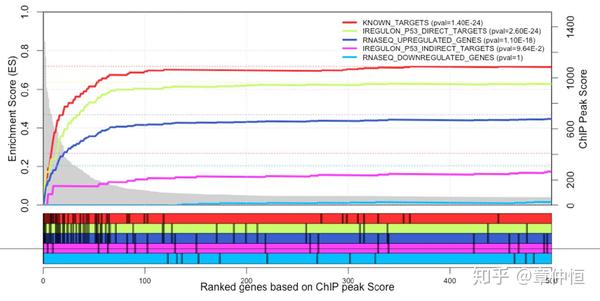

Gene Set Enrichment analysis, with on the x-axis all genes in the genome ranked according to their maximum ChIP-Seq peak (20 kb around TSS). The p53 targets (green curve) show higher enrichment than the total set of up-regulated genes (blue curve), approaching the previously known curated targets (red curve), while the non-predicted p53 targets (magenta curve) and the set of down-regulated genes (cyan curve) show no enrichment. The initial two steps in the magenta curve represent two false negative predictions of iRegulon (they fall just below the optimal cutoff), namely PLK3 and DDB2 , which are up-regulated and have a ChIP peak. P-values in the legend are calculated by the hypergeometric formula of the leading edge determined by GSEA.
GSEA分析。背景基因是ChIP-Seq获得的序列所对应的基因,让后将这些基因根据ChIP-Seq峰值进行排序,构成x轴。ChIP-Seq峰值越大说明与p53结合越紧密,也就是p53的作用位点。有排序后就可以对一个GeneSet根据Running Sum进行富集分析。毫无疑问,已知的明确的p53作用基因肯定是最容易富集的,富集分数最高的;而iRegulon预测的p53的直接目标基因也富集不错,至少比其它对照要好,说明这个开发出来的软件还是有点用的。
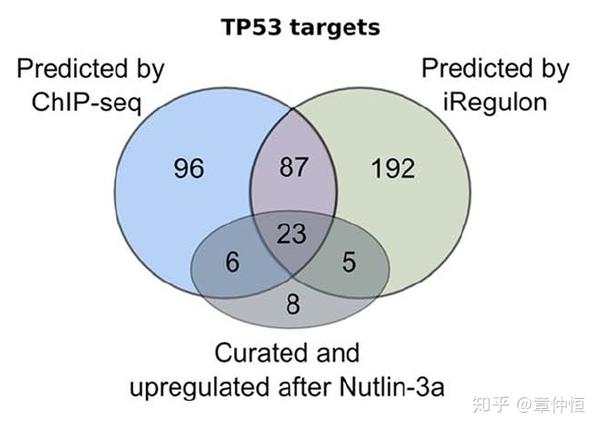
Finally, to compare how many targets are missed by iRegulon, and how many by ChIP-Seq, we again used the set of curated targets, and found comparable numbers of false negatives, namely six for iRegulon and five for ChIP-Seq ( Fig. 4C ).
反正就是用确认的基因作为p53目标基因的金标准,发现 iRegulon和ChIP-Seq假阴性结果大致相当。

In the previous section we had also found that gene repression downstream of p53 is indirect through E2F, which has been shown recently to be mediated by p21 and RB [63] , [68] . If this is true, then the down-regulated genes should not contain p53 ChIP peaks. To test this, we plotted the recovery of the 790 down-regulated genes along the p53 ChIP-peak-based gene ranking generated above ( Fig. 4B ). Similar to the indirect up-regulated genes, the down-regulated genes are completely depleted of p53 ChIP peaks (p-value = 1.0). On the other hand, the down-regulated genes are positively correlated with E2F1 ChIP-Seq data in MCF-7 from ENCODE ( Fig. S6D ).
这里先做个假设:假如p53下游被抑制的基因是通过E2F起作用,这之间又要通过p21和RB来介导;那么下调的基因就不应该包含p53的ChIP峰值。同样采用富集分析方法,结果发现下调基因在根据p53的ChIP峰值排好序的基因中基本上没有富集(只是随机出现,p=1.0)。另外也发现下调基因与E2F1的ChIP-Seq数据表现正相关。
When combining all the small p53 ChIP-Seq peaks that are detected amongst the down-regulated genes, the p53 motif is not found by de novo motif discovery, while the ChIP peaks of direct up-regulated targets are strongly enriched for de novo p53 motifs ( Fig. S6A–C ). From the ChIP-Seq validation data, we conclude that iRegulon predicts the correct master regulators (p53 and E2F) and that predicted target genes of these TFs significantly overlap with ChIP-Seq derived targets. By combining iRegulon and ChIP-Seq data, we propose a set of 110 “top targets” of p53 in MCF-7 that are directly and positively regulated. When further comparing these predicted targets to recent reports of several p53 targetomes based on combining gene expression profiles with p53 ChIP-Seq data under different experimental conditions [58] , [59] , [68] , we could confirm many common targets, but also uncovered 56 new direct p53 target genes with our analysis ( Table S6 ).
将所有有一点哪怕是很小的p53ChIP-Seq 峰的下调基因拎出来,进行重新motif发掘 [3] ,结果没发现什么。但同样的方法应用到上调基因中,却富集到了p53的motif。这里又迷糊了,一个TF对应一个motif?还是其它? de novo motif discovery(motif的重新发现)应该是指用一个PWM来计算所有序列的值,然后根据值的高低可以判定某个序列是否为p53-motif的可能性大小,不是绝对的“是”与“否”,而是一个统计分布。
这部分分析的结论是:iRegulon预测的跟ChIP-Seq获得的结果一致性较好,结合这两者的结果,有110个p53调节基因,结合最新的文献,许多位点能够再次确认,而有56个目标基因是用我们的方法新发现的。
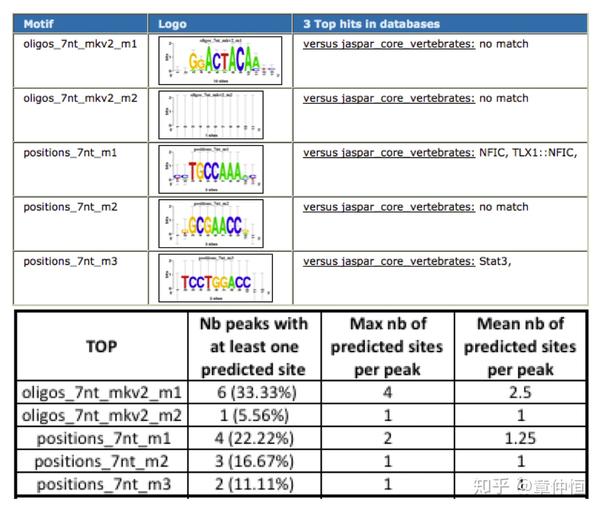
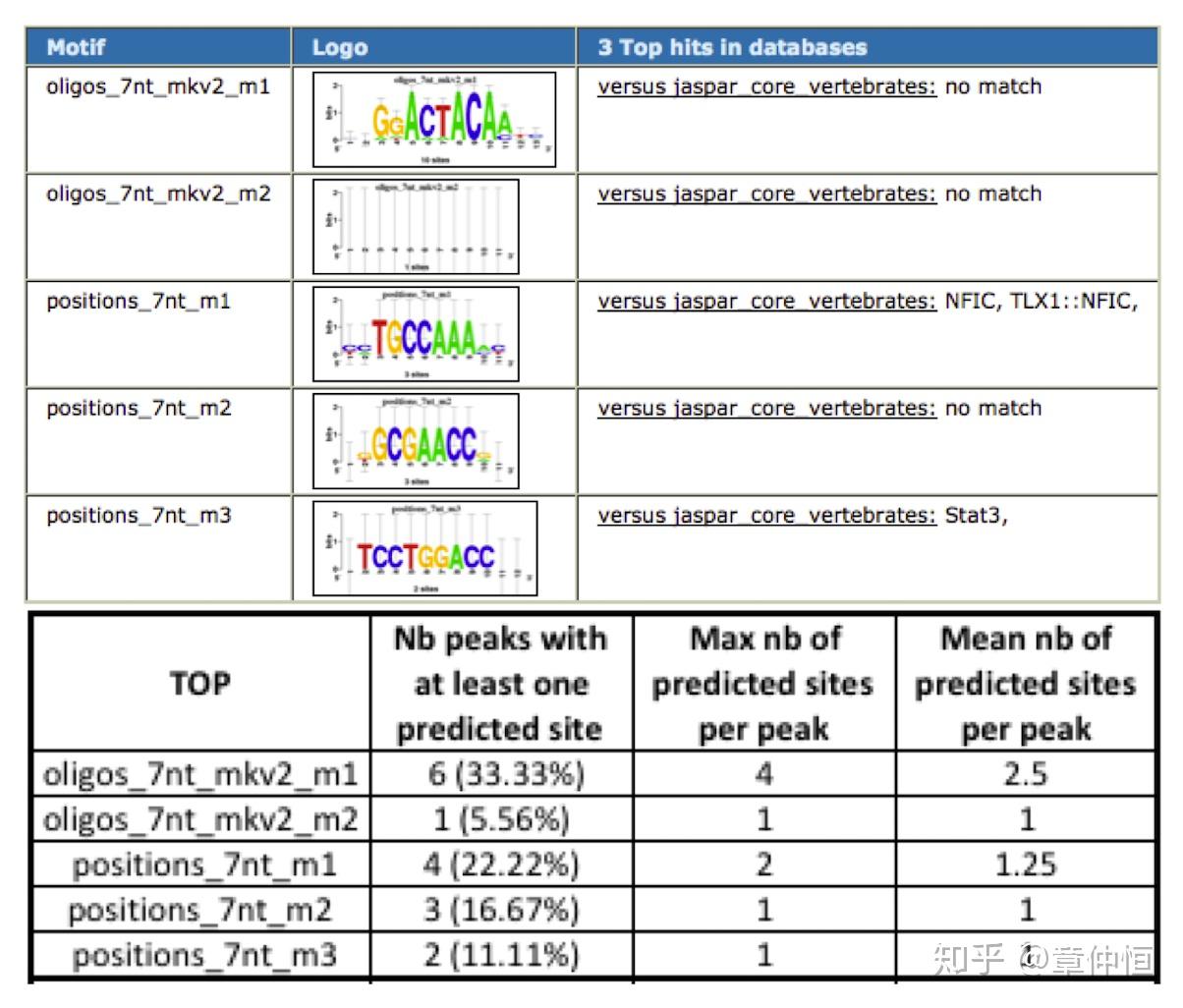
用iRegulon预测出来不是p53的直接目标基因(一个Set),可能是下调基因,这些基因周围用p53的ChiP-seq来做可能会有小的峰,这些小的峰代表了一段序列,这样的序列输入表格中motif模型,输出就是一个分数,存在一个界值,超过这个界值就可以认为是p53的结合位点,结果没有发现p53的结合位点,这些基因附近区域的序列输入motif模型,然后输出一个分数,这个分数代表了这段基因序列跟p53的亲和度。下表总共18个peaks,这些peaks中至少含有一个预测位点(用oligos_ 7nt mkv2_m1这个motif来预测)的有6个,一个peak中最多含4个位点,平均含有2.5个位点。
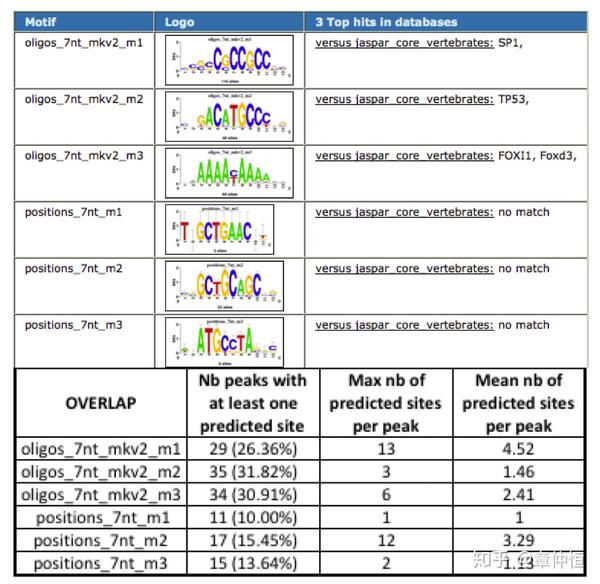

这是在上调基因中做的,在上调基因附近,选择了110个peaks,这些peaks对应的序列进行相关motif的计算,分数超过一定的值为标准,就可以预测得出结合位点。
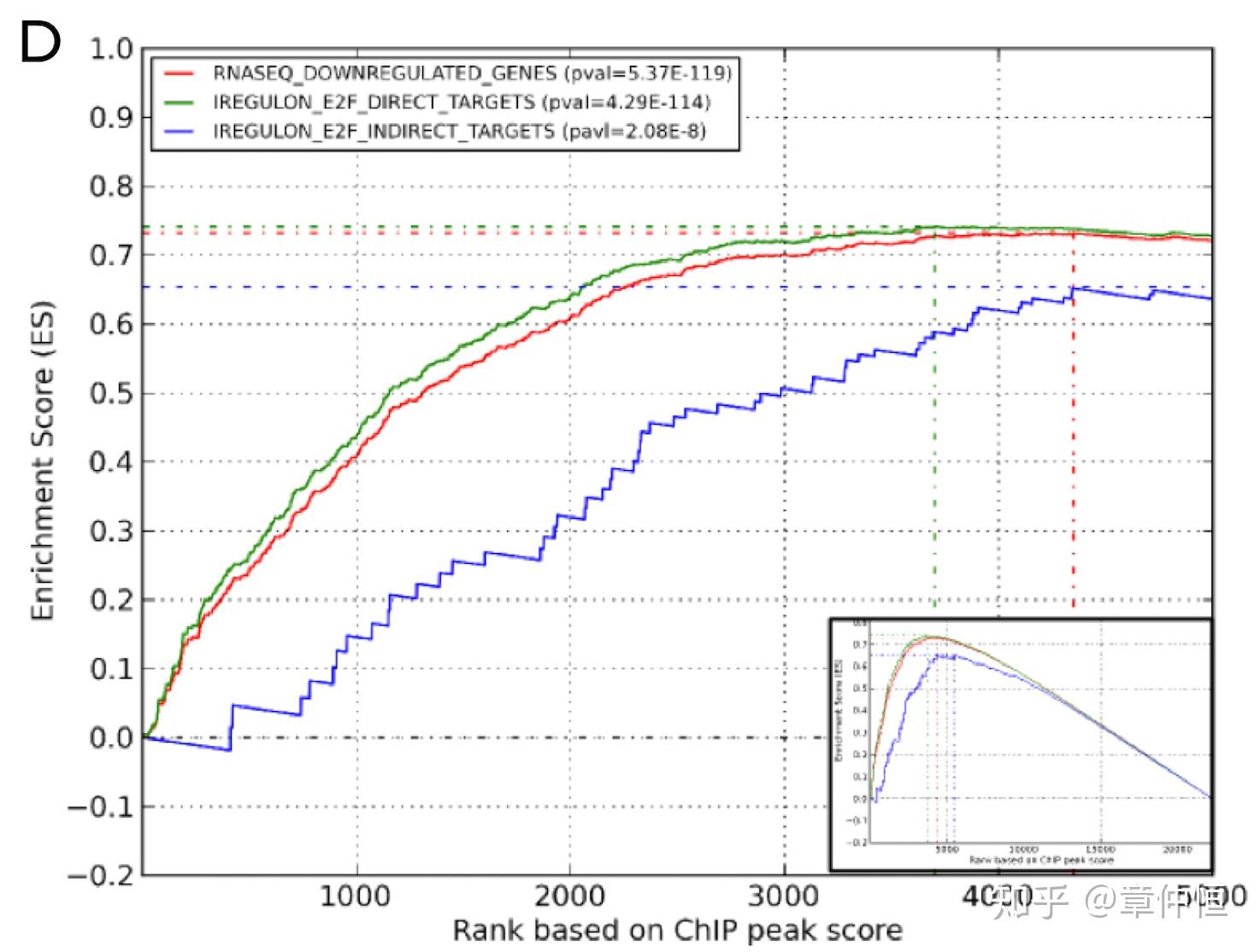
GSEA results validating the iRegulon E2F predicted targets with E2F1ChIP-Seq results. Both the total set of down-regulated genes and the predicted E2F direct targets are highly enriched. E2F ChIP-Seq data in the same MCF-7 cell line were downloaded as fastq files from ENCODE. The sequences were mapped to hg19 using same mapping parameters as for p53 ChIP-Seq experiments and the bam files of the replicates were merged with samtools.
这里做的是E2F1的ChIP-Seq结果,根据E2F1ChIP-Seq峰值可以将一系列基因排序,下调基因跟E2F预测调控基因都明显富集。
New p53 targets are confirmed by meta-analysis across human cancers and by enhancer-reporter assays
To explore the relevance of the newly identified p53 targets in other tumor types, we applied iRegulon in a meta-analysis to about twenty thousand cancer gene signatures, i.e. differentially expressed genes obtained from cancer specific experiments. We reasoned that those target genes that are recurrently predicted across cancer gene signatures, might contribute to the tumor suppressor role of p53. We used gene signatures from GeneSigDB [70] , MSigDB [71] and from gene modules generated across 91 large cancer microarray data sets (see Materials and Methods and Fig. 5A ).
新发现的p53目标基因在其它类型的肿瘤中是否有作用呢?用meta分析的方法回答这个问题,meta分析的对象是近2万个基因标签。推理:如果这些目标基因在各种肿瘤中反复被iRegulon预测,每个实验中差异表达的基因输入iRegulon,输出一个p53的regulon,每个数据集输出的回收基因可能不同,那么被反复选中的就可能参与p53的抑癌作用。文中选了几个肿瘤基因数据库。

Out of 23172 signatures, p53 is found as regulator in 709 signatures. We merged the direct p53 targets across all these signatures into a network and weighted the edges according to the recurrence of this p53-target interaction across all signatures. Many previously known p53 targets and many ChIP-Seq derived targets are recovered using this analysis (GSEA NES = 3.01, FDR<0.001) ( Fig. S7 ). Of the 110 predicted p53 targets in MCF-7 cells (as defined above), 44 are also predicted as p53 target in cancer gene signatures (grey area in Fig. 5B ).
signature就是差异表达的基因,在2万多个里有709个差异表达能富集到p53,然后建立一个网络,边的粗细就是p53-目标基因在709个网络里面出现的次数,许多已知的以及用ChIP-Seq获得的目标都被发现。在110个预测的p53目标中,44个在肿瘤基因标签中被预测出来。


Direct targets of p53 in MCF-7 cells. All genes are significantly up-regulated by p53, are predicted as p53 targets by motif discovery in iRegulon and have a significant ChIP peak. In addition, genes in the grey shaded inner circle are part of the p53 meta-regulon , meaning that they are also found as p53 targets across cancer signatures.
P53在MCF-7细胞系的直接目标用ChIP-Seq方法来发现,称为直接目标。这些直接目标对应的所有基因均上调。而这些基因用iRegulon预测也是p53作为主要调控子,同时这些target有ChIP peak。灰色区域的基因是meta分析获得的,说明这些基因在不同的肿瘤中都是p53的调控对象。
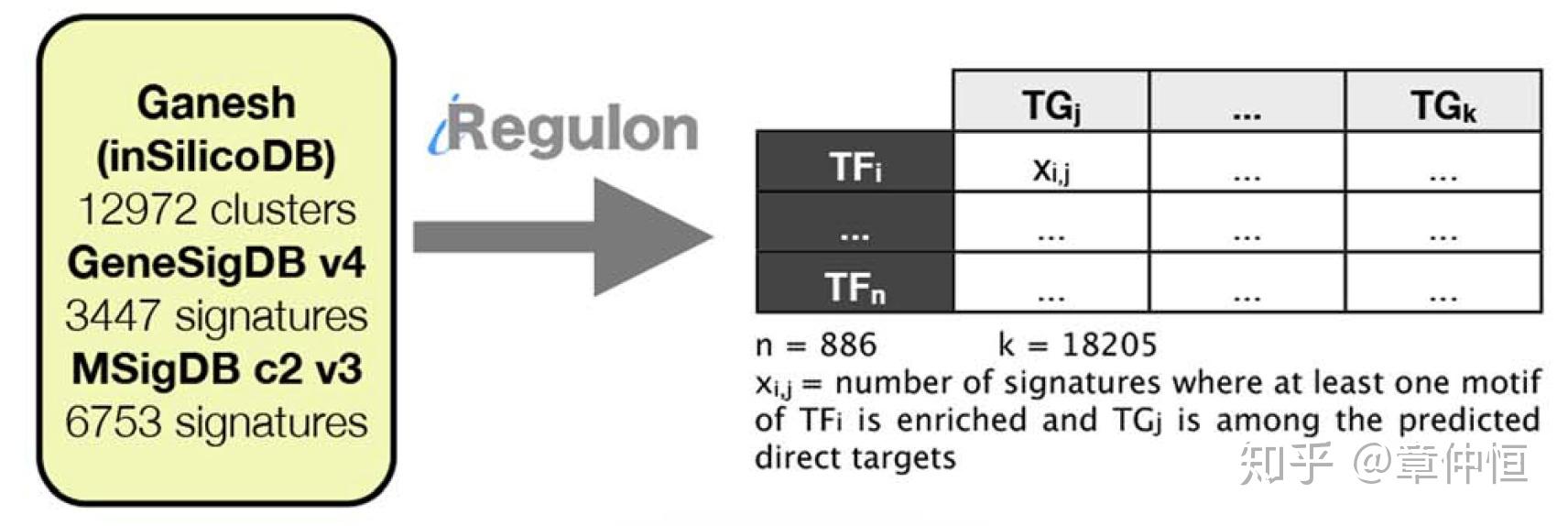
These genes are predicted as p53 targets by iRegulon and show a significant ChIP peak and are represented in the p53 cancer-related meta-regulon. Amongst these 44 genes, 20 were previously indicated as well established p53 targets (genes in squares in Fig. 5B ). When extending the analysis and including target genes recently reported in literature [58] , [59] , [68] , it becomes clear that most overlap coincides within this metatargetome (34/44) ( Table S6 ). Keeping in mind that many of the p53 targets reported by others were found using different cell lines, the enriched overlap within this metatargetome can be interpreted as a sign that these genes represent a core set targeted by p53 regardless of the cell type. Interestingly, when looking at targets like RAP2B , NHLH2 , SLC12A4 , and ALDH3A1 , they could not have been identified through motif discovery in proximal promoters only , because the p53 binding sites are located either further upstream (∼1 kb for RAP2B and ∼5 kb for ALDH3A1 ) or in introns ( NHLH2 and SLC12A4 ) ( Fig. 5C ).
灰色区域的44个基因在文献中也报告过,是确定的p53基因。值得注意的是许多其他作者报道的p53的目标基因都是来自不同细胞系的,这说明了这些是p53调控的的核心集(与细胞类型无关)。有几个基因不能通过 近处的启动区 motif discovery来发现(那前面提到的“are predicted as p53 targets by motif discovery in iRegulon”是在更广泛的区域搜寻的),这是由于这些基因的p53结合位点在上游更远的地方,或者是在内含子区域(如下图所示)。
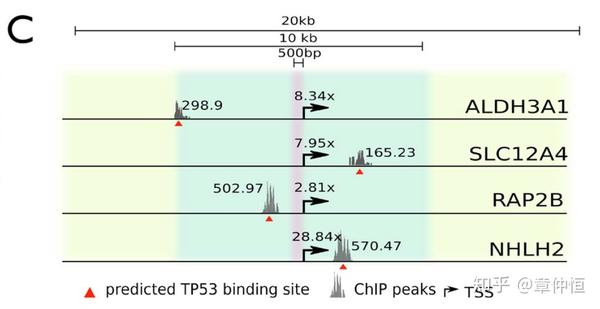

Next we confirmed experimentally whether these four targets are bona fide p53 transcriptional targets. They are all induced in a p53-dependent manner in various cellular model systems including normal diploid human fibroblasts (BJ cells) and various cancer cell lines (i.e. HCT116 and MCF-7) ( Fig. 5D ).
这四个基因实在有点特殊,所以我们要进一步实验验证一下。
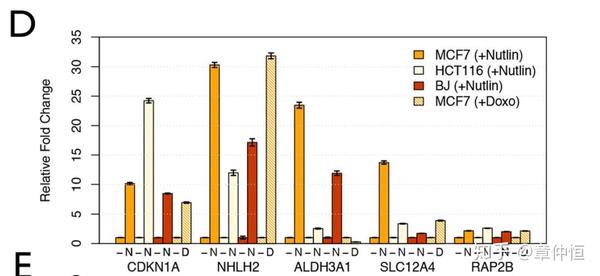

Relative mRNA expression levels of p53 target genes before (−) and 24 h after stimulation with 10 µM Nutlin-3a [4] (N) or after 1 hour pulse of 5 µM Doxorubicin (D). Expression is shown relative to non-treated control and normalized to optimal reference genes for each cell type, assessed by GeNorm [130] . Error bars show standard error of the mean (SEM) of 3 replicates.
Except ALDH3A1 , they are also all significantly induced upon exposure to the DNA damaging agent doxorubicin, a well-established p53 inducer (adjusted p-value<0.05). Their kinetic of induction both in response to Nutlin-3a and DNA damage is comparable to the one seen with known direct p53 targets such as CDKN1A further supporting a direct role for p53 in their regulation ( Fig. S8 ).
Doxorubicin是个DNA破坏剂,处理以后p53介导的基因表达量均增高。p53启动修复。而Nutlin-3能激活p53,所以与Doxorubicin的功效是一样的。 CDKN1A基因是已知的p53作用点,而其它四个与CDKN1A表现相同,说明这四个都是p53的作用点。
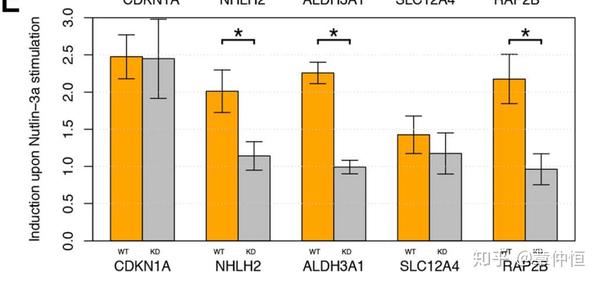

Finally, for all except one we could confirm luciferase reporter activity of the predicted p53 enhancer region ( Fig. 5E ). Enhancer-reporters for ALDH3A1 , NHLH2 and RAP2B show a significant induction after Nutlin-3a treatment in wild type but not in a p53 knock-down (KD) cell line (p-value<0.05). SLC12A4 does not have a significant induction in either cell-type. Note that our positive control enhancer, namely the CDKN1A promoter, is a very responsive p53 target and likely responds to low levels of p53, which could explain the induction that is still observed even under p53 KD conditions. Functionally, these validated p53 target genes have been implicated in p53-regulated processes such as the control of cell volume, growth and movement (SLC12A4 and RAP2B) and metabolism (ALDH3A1 and NHLH2).
p53敲除的细胞用Nutlin-3a处理后, luciferase reporter活性显著降低。换句话说,除了 SLC12A4外,其它细胞的野生型在 Nutlin-3a处理后Enhancer-reporters显著增高。 CDKN1A是一个阳性对照组,其对p53作用目标非常敏感,非常低的p53也能导致其激活。但问题是,既然p53敲出了,应该是一点也p53也没有,剂量绝对为0的,这里不太明白。
Motif and track discovery join forces
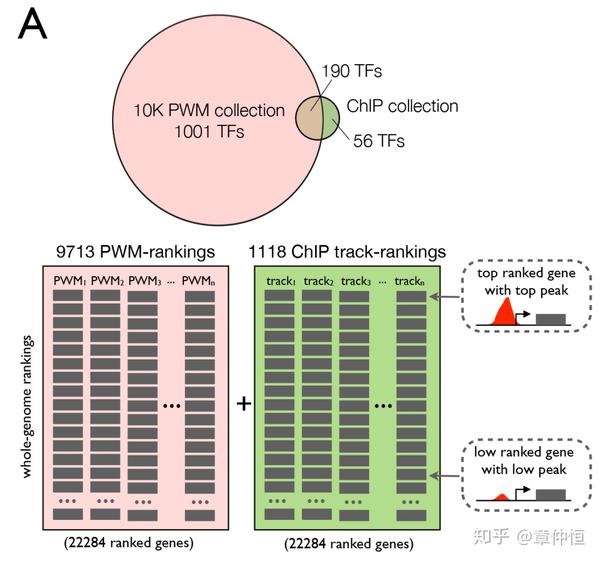
We extended our motif discovery approach to allow the discovery of significantly enriched ChIP-Seq tracks in a set of co-expressed genes. We created a database with track-based gene rankings from a collection of 1118 ChIP-Seq experiments against 246 human sequence-specific TFs across 40 cell types and apply the same “ranking-and-recovery” enrichment calculation as employed earlier (see Materials and Methods ). These and other recent resources further enlarged our motif collection to 9713 distinct PWMs (“10K collection”) ( Table 1 ). To test whether motif and track discovery can be performed simultaneously, we combined the motif-based rankings and the track-based rankings into one enrichment analysis, although each AUC score distribution is kept separate for normalization ( Fig. 6A–B ).
此部分内容将扩展了motif发现的,motif其实是对一系列序列的定量总结,对motif的扩展就是对序列的扩展。但是后面谈到扩展的方法是将ChIP-Seq tracks限制在一套共表达基因上,相比于全基因组的ChIP-Seq,这里似乎是缩小了范围。怎么破?还是需要进一步了解一下ChIP-Seq tracks的工作方式。其实这里可能是说细胞类型这些增加了,而共表达基因不是来限制ChIP-Seq实验的(查看ChIP-Seq峰的时候只能限定那几个共表达的基因附近),而是用来预测TF Regulon的。因此结合下文,这里的“extend”是指数据库的扩展,这里已经扩展到了9713个PWM。
这里作者建立了一个数据库,包含246个TF,用了40种细胞类型,做了1118个ChIP-Seq实验,这样理解起来1个TF可能有几种motif(PWM),因为细胞不同。然后用“排序-回收”的方法对这些共表达基因进行富集。
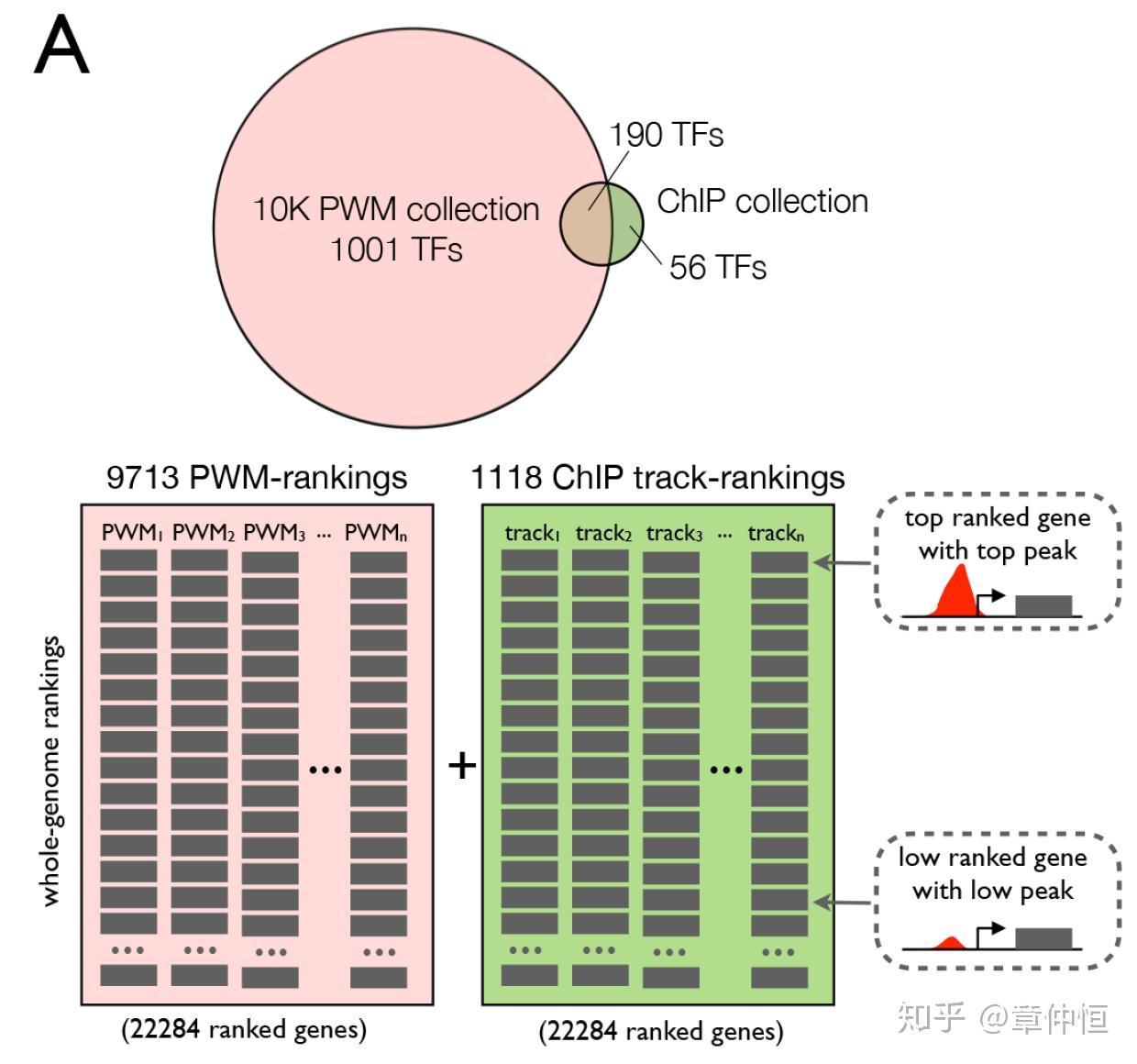
PWM排序和track排序其实是用不同方法做同一个事情。PWM排序根据给定基因附近序列的ATCG计算出得分,而trank法根据基因对应的峰值将基因排序。其实很大程度上,峰值越大其PWM评分也越高,但由于各种技术原因,这两者不是一一对应的关系,因此两者互相验证能使结果更加可靠。有了这个排序之后就可以进行回收,计算AUC值。

p53标签就是指p53扰动后差异表达的基因列表,这个列表分别输入到上述两个排序中,就可以给出AUC最大的那个多对应的PWM或track。
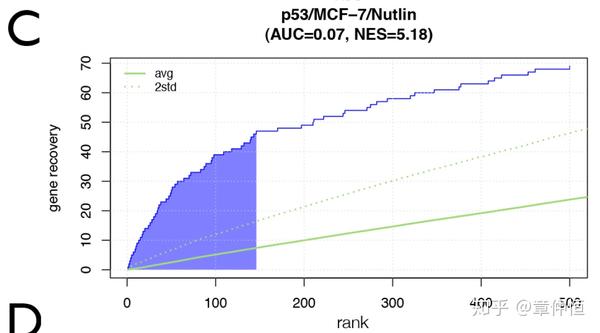

这里其实是截图的一部分,横坐标应该是22284个基因,默认的前3%就是 22284\times3\%=668 ,这里似乎只有150个基因,那是 150/22284=0.0067 。但不管怎样,比随机回收率(绿色线)是高多了,甚至显著高于其2倍的标准差。
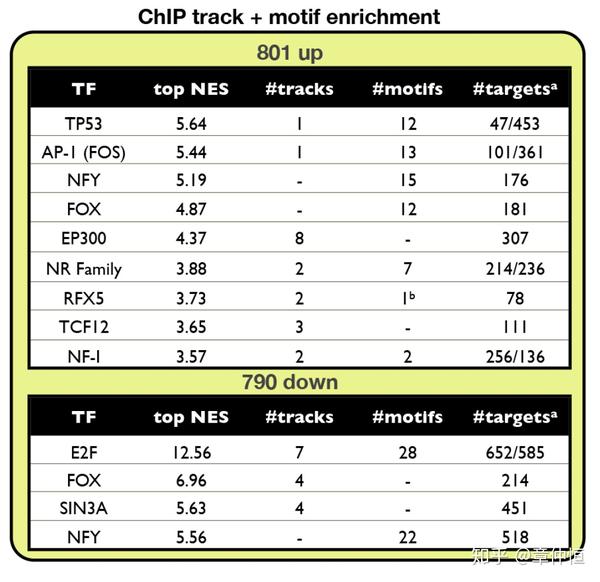

以为有不同的实验、不同的细胞、不同环境,一个TF下去做ChIP,可能出来不同的结合位点和峰值,这时候就有不同的PWM或者track,图例说明里说NES=2.82,表里显示的是NES=3.73。
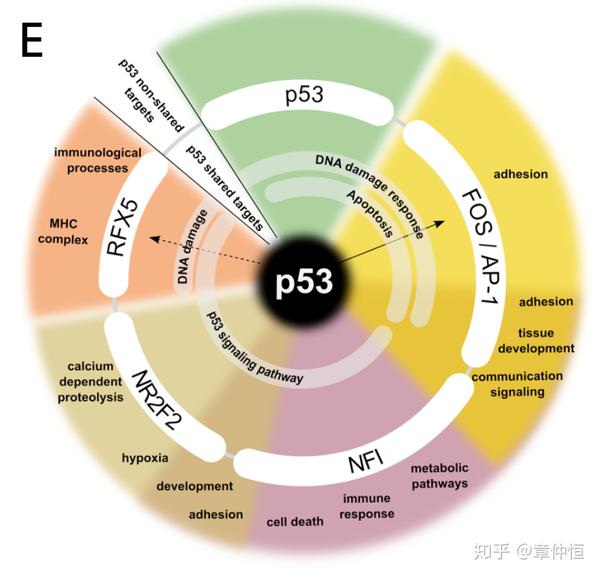

Functional categories found enriched for predicted co-factors of p53. The annotation of p53-shared targets is shown in the inner circle, while the annotation of non-shared targets (for example, AP-1 targets but not p53) is shown on the outer circle. The co-factors shown here are those found by both motif and track enrichment.
这些功能富集主要是在目标基因(targets)上进行的,将每个TF的目标进行GO或KEGG富集,就能得到一些功能标注,这些功能标注有些是共有的,有些是非共有的,这个 circo图 就能很好的表示了。
Discussions
We have optimized and expanded motif discovery methods and used large collections of up to 10.000 candidate motifs to facilitate translation of motif detection results into a network biology framework. By adding this network-layer on top of cis -regulatory motifs, we could generate direct insight into a biological process, rather than producing a mere list of enriched motifs from a gene set. iRegulon outperformed existing methods at detecting the correct upstream regulator. We found that using PWMs from other species than human greatly helps motif detection in human data sets. Many TFs are conserved from human to mouse, and even from human to fly or yeast, and sometimes the yeast or fly PWM is of higher quality or better captures the specificity of DNA binding. In addition, we found that using multiple PWMs for the same TF is an advantage and leads to higher performance of TF recovery compared to using non-redundant motif collections. Our motif collection also contains an important fraction of “novel” motifs for unknown TFs. These motifs are mostly derived from whole-genome computational predictions. In some cases these unknown motifs are clustered together in the output of iRegulon alongside a known motif, and can thereby lead to candidate TF predictions, while in other cases they may represent orphan motifs (with unidentified TFs). The mixture of known and unknown motifs creates a hybrid motif detection approach, combining de novo motif discovery and pattern matching approaches.
讨论部分读一下也很有必要,能够梳理本文所写的内容,以及与其它背景知识相互间的关系。本文主要优化扩展了motif发掘方法,用10k的motif将motif发掘(排序-回收方法)的结果转化为生物网络模型。顺式调控元件上加上网络层能够帮助更好理解生物学行为,而不仅仅是从一批共表达基因中富集得到一系列motif。iRegulon在发掘上游调控子方面比其它方法要好。我们也发现用利用其它物种的PWM能辅助人类的motif发掘。许多TF在任何小鼠中都是高度保守的,而在一些低等动物如果蝇等的PWM更加高质量(更好地反应TF与DNA结合的特异性,可能与控制得更好的实验条件有关)。
另外我们也发现,对于给定的TF用多个PWM有一定的优势:TF回收更加高效。多个PWM也就是多个motif,一个TF会有很多个PWM-ranking,如果我们选择其中回收最高(NES值最大)的一个,那势必会增加TF的富集,任意一套基因更有可能发现motif,这称之为提高performance。本研究的PWM库里也有一些未知TF的motif,这些主要是由计算机预测的;根据本文的知识,这部分是不太好理解了。按照常理motif都是根据给定TF的ChIP-seq来构建的,而由计算机直接预测是什么鬼?在iRegulon结果输出中,有时候这些motif跟已知TF的motif聚类在一起,这样就可以把这些已知的TF当成是未知motif的对应TF,从而实现了一次预测。有时候这些计算机预测的未知TF可能是孤儿motif。这样将已知和未知TF的motif结合起来的方法叫杂交motif发掘,技术上包括了motif的重新发现(根据iRegulon的聚类输出)和类型匹配。
Large-scale analyses of co-expressed gene sets of different origins, including co-expression, TF binding (ChIP), protein-protein association networks and microRNA targets, suggest that by exploiting the genome sequence, together with other species' genomes and collections of consensus TF binding sites, the most relevant sub-networks that underlie observed changes in gene expression or observed genetic interactions can be reconstructed. In up to 70% of the cases, the upstream regulatory factor can be identified, along with a set of direct targets. Therefore iRegulon provides an alternative approach to probe a particular biological process when gene expression data is available but the TF is not known in advance and/or ChIP-Seq is not feasible. By combining iRegulon with RNA-Seq, the resolution of gene expression profiling and gene regulatory network mapping can be increased, allowing the characterization of any cell type, cellular response, or tumor sample, up to the single cell level.
共表达基因的来源很多,包括了共表达、TF结合、蛋白-蛋白关联网络以及mRNA作用目标基因。通过对基因组序列的分析,参考比对其它物种的基因组,以及已形成共识的TF结合位点,就能够构建与基因表达及遗传交互最相关的子网络。在70%的案例中,能够识别上游转录因子,以及一套直接作用目标基因(这应该是目标一套基因的一个子网络)。在这里,iRegulon的作用就是在已知基因表达数据,但TF未知或者ChIP-Seq不能做的的时候,去探索一个生物学过程。通过结合iRegulon和RNA-Seq,基因表达特征及基因调节网络绘制的分辨率会增高,能看清更加细节的东西。这就使得研究细胞类型、细胞反应或肿瘤样本,甚至单细胞水平都成为可能。
Multiple regulons are often discovered from one co-regulated gene set. This is expected because in higher vertebrates gene regulation is combinatorial, where multiple TFs cooperate, either through binding in the same CRM (called heterotypic CRMs), or in separate CRMs of the same target gene [17] . In addition, the targets of a TF can be TFs themselves, and in turn activate or repress their own targets. For example, in the p53-dependent gene set iRegulon identified not only p53 as regulator, but also a previously known co-factor AP-1 and new regulators downstream of p53 such as RFX5. Interestingly, FOS and FOSL1 , important members of the AP-1 complex, and RFX5 , were all identified in this study as targets of p53. These regulators can explain a large proportion of the possible target genes of p53 as being indirect and regulated by another TF. When we extended our ranking-and-recovery framework to include more than one thousand ChIP-Seq data tracks, we also found the respective ChIP-Seq peaks for AP-1, RFX5, and several other co-factors as significantly enriched in the p53 downstream network. The joint finding of both a motif and a track for the same transcription factor strongly increases the confidence for these factors to play a role in the network as master regulator (i.e., directly controlling many target genes). Nevertheless, we envision that in most cases the motif enrichment alone, without any track enrichment, can directly lead to candidate master regulators, because ChIP-Seq data is condition-specific and is currently available for relatively few transcription factors.
一般能从一套共表达基因中发掘多个转录子。这应该是合理的,因为在高等脊椎动物中基因的调节是多因素的,往往是多个TF共同协调,可能是通过结合到相同的CRM,或者同一个基因不同的CRM来实现。另外,TF的目标基因可能是其它的TF,通过这样作用就可以进一步激活或抑制下游目标 。例如本研究所展示的,p53依赖的一套基因发掘出的调节子不仅仅包括p53调控单位,还包括了之前实验已知的共协同因子AP-1,以及其它p53下游的调节子(如RFX5),有趣的是,AP-1复合体的重要组分 FOS和FOSL1,以及 RFX5都被发现是p53的下游调控目标。这些调节子能够解释p53的大量的目标基因是通过间接作用起作用,间接意味着中间还有其它的TF。排序-回收法主要用在PWM库里,但我们将此扩展到ChIP-Seq data tracks时,我们也观察到了AP-1和RFX5的ChIP-Seq峰,以及其它一些共同调节因子,富集与p53下游网络。这里master regulator的定义终于搞清楚了,主要是 直接 调控许多目标基因的TF。共表达基因【用“排序-回收”法】找到的motif和track对应了同一个TF,这就增加了这些因子(motif和track都对应的TF)作为master regulator的可信性。motif其实是通过ChIP-seq的结果算出来的,那跟用一个ChIP-seq轨道得出的原理差不多吧。motif是PWM,是计算机对TF跟某基因序列片段结合亲和度的模拟;而track是直接实验,看TF跟基因序列片段结合的亲和度(track的峰值);一个是计算机模拟,一个是直接实验证明,如果两者都一致,那确实能说明该TF是这批基因的master regulator(大师调节子),必须是直接调控。但作者认为,大部分情况下,发掘master regulator只要做motif富集就够了,因为ChIP-Seq数据在不同实验条件下会有不同 ,而且只有很少一部分TF会有ChIP-Seq数据。但这里问题来了:不是说motif数据其实也是根据ChIP-Seq数据算出来的吗?
The absence of a regulator in the output of iRegulon, when neither a motif nor a track is enriched, can also be informative. For instance, neither the p53 motif nor its ChIP-Seq track are found enriched among the down-regulated genes, leading to the hypothesis that p53 does not act as a direct repressor, but only as an activator. Rather, iRegulon points to E2F as the master regulator of the down-regulated genes, both by its motif and track. This finding can be explained as indirect down-regulation of E2F targets and has recently been experimentally established: p21 controls RB1-mediated repression of E2F targets, including E2F family members themselves, thereby reinforcing this signal further [63] , [68] .
iRegulon分析结果提示没有motif或track被富集,这本身也是个很有价值的信息。例如,在本研究的下调基因中,p53的motif和track都没有富集,我们就可以假设p53不是个直接的抑制因子,但是作为激活因子。iRegulon分析提示E2F是下调基因的大师调节子(motif和track都证实)。根据这些观察结果推测,p53先激活E2F,E2F再抑制基因表达,这个推测已经被实验证实。p21控制着RB1接到的E2F目标基因下调,包括E2F家族成员。
Our experimental findings on the p53 regulon were obtained in MCF-7 breast cancer cells. Usually, one iRegulon analysis is focused on one biological process, and predicts transcriptional targets that are relevant in that particular cell type or condition under study. We show that it is also possible to apply iRegulon more systematically on multiple signatures to identify cancer-related ‘meta-regulons’. They often represent the canonical, high-confidence target genes and agree well with ENCODE ChIP-Seq data ( Fig. S7 ). This shows that relevant TF-target interactions can be identified purely from the genome sequence, thereby creating a valuable resource for less studied TFs.
我们研究p53调节模块主要是根据MCF-7乳腺癌细胞。通常而言,iRegulon分析主要集中在一个生物学过程,并预测那个条件下的转录目标。我们的结果也显示,我们可以用iRegulon更加系统地分析多个基因标签(各种不同的肿瘤中进行p53干扰后产生的差异表达基因),从而获得多种肿瘤相关的“后调节模块”,这些模块都是经典的、高可信度的目标基因,跟ENCODE ChIP-Seq数据非常一致。TF-目标之间的相互作用能从基因序列中获得,这对于研究较少的TF能提供更多的资源。
参考
- ^ ChIP-Seq identifies the binding sites of DNA-associated proteins and can be used to map global binding sites for a given protein. ChIP-Seq typically starts with crosslinking of DNA-protein complexes. Samples are then fragmented and treated with an exonuclease to trim unbound oligonucleotides. Protein-specific antibodies are used to immunoprecipitate the DNA-protein complex. The DNA is extracted and sequenced, giving high-resolution sequences of the protein-binding sites.
- ^ 就是指那115个给定的TF
- ^ De novo motif discovery allows you to directly query the sequence to discover which motifs are the MOST enriched sequences in your target set. Known motif discovery will simply tell you which of the known motifs is most enriched in your target set.
- ^ Nutlin-3 is a specific MDM2 inhibitor, which blocks the p53-binding domain of MDM2 resulting in p53 stabilization and p53 activation
