


深度生成模型:重塑药物发现的未来【2】工具箱:大型生物医学数据集、化合物/分子的表示

深度生成模型:重塑药物发现的未来【 @金平宇 】
【2】深度生成模型的工具箱:用于药物发现的大型生物医学数据集、化合物/分子的表示
【3】深度生成模型的工具箱:RNN / VAE / GAN / Flow-based / RL
深度生成模型的工具箱
设计一种新药是一个复杂的任务,需要满足预先定义的一系列标准,包括目标位点的效力、与非目标位点的特异性、物理特性以及其他化学和生物学指标等。传统方法需要化学家从广泛的化学空间中选取并通过实验验证候选分子,效率较低。深度生成模型因其能够在节约时间和成本的同时自动生成具有生物活性和可合成性的新分子而备受青睐。
用于药物发现的大型生物医学数据集
我们首先简要概述了几个常用的化学和生物信息学数据库,它们提供标记和未标记数据以用于训练、验证和测试深度生成模型,以服务于药物发现社区。制药公司拥有自己的专有化合物收藏,规模在2-3百万个化合物,并伴随着从过去的药物发现任务中收集到的相关数据。在公共领域中,ZINC数据库收集了近20亿种可购买、商业化“类药物”化合物以进行计算机筛选。 [1] 由于其巨大的规模,它也可用于为预训练生成模型学习分子模式。具有生物活性的分子,如在手动筛选的ChEMBL数据库中所包含的近150万个真正具有生物活性的分子,每个分子都至少具有一个实验生物活性测量值, [2] 尤其值得关注。它们可用于训练模型以生成具有特定属性的分子。GDB-17数据库 [3] 列举了大多数有机分子(1664亿种)的17个重原子的C、N、O、S和卤素化合物,包括许多低分子量小分子药物以及更小的典型前导化合物。超大型化学数据库, [4] 如Enamine( Home - Enamine )和REALdb, [5] 包含数十亿个可合成的化合物,由化学信息学方法和专家系统类型规则鉴定。这些超大型数据库为培训具有广泛适用性的模型提供了机会。除小分子资源外,几个大分子数据库也为大分子设计的生成模型培训提供了丰富的数据,如PDB数据库。 [6]
化合物/分子的表示
分子的表示对于生成模型非常重要。有三种类型的表示:(1)基于序列的,(2)基于图形的,和(3)图像(图2)。自然语言处理(NLP)的巨大成功启发了一种类似于人类语言的方式来描述分子中的符号。生物结构中的语义和语法类似于人类语言;因此,分子可以表示为字符序列。新型的小分子设计通常使用简化的分子输入行输入系统(SMILES)。 [7] 基于序列的结构是通过遵循SMILES语法规则编码到向量中生成的(图2A)。更直接的表示分子的方法是基于图形的。 [8] 在图形表示中,小分子的原子形成节点集,键被视为边(图2B)。对于大分子,接触图 [9] 是表示任意两个氨基酸残基对之间距离的图形。在大量节点上训练基于图形的模型是昂贵的,因为空间复杂度随节点数量的平方而增加。 [10] 与基于序列的方法相比,基于图形的表示易于实现为图形卷积层,并且键权重可以在消息传递网络中进行优化。基于序列的表示通常是紧凑、内存效率高和易于搜索的。然而,基于序列和基于图形的方法都无法捕捉与生物学有关的配体-蛋白质相互作用中配体或蛋白质的三维信息。分子的三维构象捕获了原子的相对方向 [11] [12] [13] [14] (图2C)。最近也提出了几种最新的三维表示。 [15] [16] [17] DEVELOP结合了现有的基于图形的深度生成模型De-Linker,以及卷积神经网络来利用分子和目标药效团的三维表示。 [18] DeepLigBuilder是一种基于图形的生成模型,利用配体-受体相互作用的三维结构表示,进行端到端的设计,生成具有药物相似性特性的化学和构象有效的三维分子。 [19] 传统的蛋白质图像或三维表示需要从冷冻电镜和晶体学获得准确的三维结构数据,这很难实现。最近的AI方法,如AlphaFold2,可以提供大量蛋白质三维数据来应对这些挑战。 [20]

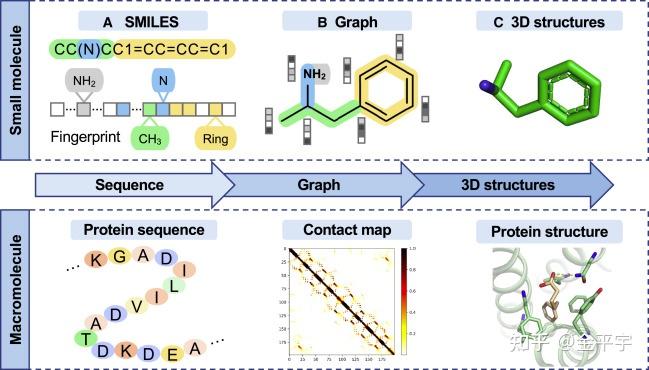
三种分子表示方法包括:(A)一维(1D)基于序列的表示方法;(B)基于图的表示方法;(C)适用于小分子和大分子(即蛋白质)的三维表示方法。接触图矩阵的值为1,如果距离大于预定阈值,则为0。
Reference :
- Zeng, X., Wang, F., Luo, Y., Kang, S.G., Tang, J., Lightstone, F.C., Fang, E.F., Cornell, W., Nussinov, R. and Cheng, F., 2022. Deep generative molecular design reshapes drug discovery. Cell Reports Medicine , p.100794.
参考
- ^ Irwin J.J., Tang K.G., Young J., Dandarchuluun C., Wong B.R., Khurelbaatar M., Moroz Y.S., Mayfield J., Sayle R.A. ZINC20-A free ultralarge-scale chemical database for ligand discovery. J. Chem. Inf. Model. 2020;60:6065–6073.
- ^ Gaulton A., Bellis L.J., Bento A.P., Chambers J., Davies M., Hersey A., Light Y., McGlinchey S., Michalovich D., Al-Lazikani B., Overington J.P. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012;40:D1100–D1107.
- ^ Ruddigkeit L., van Deursen R., Blum L.C., Reymond J.L. Enumeration of 166 billion organic small molecules in the chemical universe database GDB-17. J. Chem. Inf. Model. 2012;52:2864–2875.
- ^ Patel H., Ihlenfeldt W.D., Judson P.N., Moroz Y.S., Pevzner Y., Peach M.L., Delannée V., Tarasova N.I., Nicklaus M.C. SAVI, in silico generation of billions of easily synthesizable compounds through expert-system type rules. Sci. Data. 2020;7:384.
- ^ Hoffmann T., Gastreich M. The next level in chemical space navigation: going far beyond enumerable compound libraries. Drug Discov. Today. 2019;24:1148–1156.
- ^ Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. The protein Data Bank. Nucleic Acids Res. 2000;28:235–242.
- ^ Weininger D. A chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Model. 1988;28:31–36.
- ^ Schwalbe-Koda D., Gómez-Bombarelli R. Machine Learning Meets Quantum Physics. Springer; 2020. Generative models for automatic chemical design; pp. 445–467.
- ^ Gupta N., Mangal N., Biswas S. Evolution and similarity evaluation of protein structures in contact map space. Proteins. 2005;59:196–204.
- ^ David L., Thakkar A., Mercado R., Engkvist O. Molecular representations in AI-driven drug discovery: a review and practical guide. J. Cheminform. 2020;12:56.
- ^ Gainza P., Sverrisson F., Monti F., Rodolà E., Boscaini D., Bronstein M.M., Correia B.E. Deciphering interaction fingerprints from protein molecular surfaces using geometric deep learning. Nat. Methods. 2020;17:184–192.
- ^ Wójcikowski M., Kukiełka M., Stepniewska-Dziubinska M.M., Siedlecki P. Development of a protein–ligand extended connectivity (PLEC) fingerprint and its application for binding affinity predictions. Bioinformatics. 2019;35:1334–1341.
- ^ Mahmoud A.H., Masters M.R., Yang Y., Lill M.A. Elucidating the multiple roles of hydration for accurate protein-ligand binding prediction via deep learning. Commun. Chem. 2020;3:19.
- ^ Jones D., Kim H., Zhang X., Zemla A., Stevenson G., Bennett W.F.D., Kirshner D., Wong S.E., Lightstone F.C., Allen J.E. Improved protein–ligand binding affinity prediction with structure-based deep fusion inference. J. Chem. Inf. Model. 2021;61:1583–1592.
- ^ Xu M., Wang W., Luo S., Shi C., Bengio Y., Gomez-Bombarelli R., Tang J. International Conference on Machine Learning. PMLR; 2021. An end-to-end framework for molecular conformation generation via bilevel programming; pp. 11537–11547.
- ^ Shi C., Luo S., Xu M., Tang J. International Conference on Machine Learning. PMLR; 2021. Learning gradient fields for molecular conformation generation; pp. 9558–9568.
- ^ Axelrod S., Gómez-Bombarelli R. GEOM, energy-annotated molecular conformations for property prediction and molecular generation. Sci. Data. 2022;9:185–214.
- ^ Imrie F., Hadfield T.E., Bradley A.R., Deane C.M. Deep generative design with 3D pharmacophoric constraints. Chem. Sci. 2021;12:14577–14589.
- ^ Li Y., Pei J., Lai L. Structure-based de novo drug design using 3D deep generative models. Chem. Sci. 2021;12:13664–13675.
- ^ Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Žídek A., Potapenko A., et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–589.
