基因组规模上的聚合数据类型的相似性网络融合
近期的技术已经使收集不同类型的全基因组数据十分划算,结合这些数据去创建一个给定的疾病或生物过程的一个全面视图的计算方法是有必要的。相似网络融合(SNF:similarity network fusion)通过创建每一个可利用的数据类型的样本(如,患者)的网络可以解决这个问题。例如,创建一个给定一群患者疾病的全面视图,SNF计算并融合分别来自于他们每一个数据类型的患者相似网络,目的是利用数据中的互补性。我们使用SNF去结合五种癌症数据集中的的mRNA表达,DNA甲基化和microRNA(miRNA)表达数据。SNF算法大大优于单一数据的分析与建立的综合方法,这个优势在识别肿瘤亚型和预测生存时是有效的。
一、SNF算法流程
图1.SNF步骤示例图
其中,图1a是来自同一类患者的mRNA表达和DNA甲基化;图1b是对于每一个数据类型的患者-患者相似度矩阵;图1c是患者-患者相似网络,节点代表患者,边代表一对患者间的相似度;图1d是网络融合过程,通过SNF算法迭代地通过其他网络的信息来升级每一个网络,使其每一步更相似;图1e是交互网络融合导致集合为最终融合网络,边的颜色表示数据类型已经贡献到给定的相似度。
二、SNF算法模型
2.1 网络权值(边)设置(患者相似度W(i,j))
假设有n个样本(例如,患者数据),m个测量值(例如,mRNA基因表型)。
G=(V,E) :患者相似度网络。顶点V表示患者{x1,x2,...,xn},边E是权重表示患者相似度。
ρ(i,j) :患者xi与患者xj的欧几里得距离
W(i,j) :患者xi与患者xj的相似度矩阵(n×n)
其中患者相似度W(i,j)使用比例指数相似核(
scaled exponential similarity kernel
)定义:
其中,μ是一个超参数,可以通过经验去设置,推荐设置的范围为[0.3,0.8];εij是用于消除缩放比例问题,这里定义εij为:
其中 mean(ρ(xi,Ni))是xi到每一个邻居的距离均值。
2.2 权值矩阵标准化
为了计算来自多重测量值类型的融合矩阵,我们在定点集V上定义一个全稀疏核(full and sparse kernel),将其标准化后的权值矩阵为
 。
。
其中D为一个对角矩阵。
且
 成立。
成立。
然而这种标准化可能造成数值的不稳定,因为其涉及在对角线上自相似的W。
一种更好的标准化的方法如下:
这样的标准化可以去除对角线上自相似的尺度,且
仍然成立。
2.3 K个最相似患者的相似度S(i,j)
设Ni表示xi的邻居(包括xi)在网络G中。
给定一个图G,我们使用K最邻近的方法(KNN)去测量局部亲和力(local affinity)得到Ni,并得出相邻点之间的相似度:
其中,非相邻点的相似度为0.
请注意,P(i,j)是第i位患者与其他所有患者的相似性,而S(i,j)是第i个患者与其k个最邻近患者的相似度!
在算法中,总是把P(i,j)作为初始状态,而S(i,j)作为核心矩阵在捕获局部结构和计算效率的两种容量的融合过程中。
2.4 相似性网络融合(SNF)
设有m个不同的数据类型,对于第v个数据类型(v=1,2...m),通过式(1)得出对应的相似度矩阵为
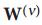 ,并分别通过式(2)与式(3)计算出
,并分别通过式(2)与式(3)计算出
 与
与
 。
。
我们以两个数据类型为例,即m = 2.
Step1.计算数据类型的P(i,j)与S(i,j)。
通过式(2)计算出
 与
与
 ;通过式(3)计算出
;通过式(3)计算出
 与
与

让
 与
与
 分别代表当t=0时两个初始状态矩阵(t表示时间)。
分别代表当t=0时两个初始状态矩阵(t表示时间)。
Step2. 迭代更新相似度矩阵。
这是SNF的关键步骤,分别迭代更新每一个数据类型的相似度矩阵,如式(4)(5):
其中
 是第一个数据类型的状态的 t 步后的状态;
是第一个数据类型的状态的 t 步后的状态;
 是第二个数据类型的状态的 t 步后的状态。
是第二个数据类型的状态的 t 步后的状态。
在 t 步后,整体状态矩阵可以这样计算:
然而S是一个KNN图,它可以降低距离间的噪音,于是,另一种迭代更新方法是将公式(4)升级为如下公式(6):
其中,Ni代表着包括节点xi的xi的邻居数。
通过相同的方法,我们也可以升级公式(5)为与公式(6)类似的公式
Step3.相似网络融合
将Step2中得到的
 与
与
 代入公式(2)中进行标准化后,就得到了融合后的各个数据类型图中边上的权值信息。
代入公式(2)中进行标准化后,就得到了融合后的各个数据类型图中边上的权值信息。
广泛地,当m>2时,公式(4)(5)便成了如下公式(7):
三、网络聚类(以疾病子类型为例)
3.1 划分矩阵的设置
给定n个样本和m个测量值,我们的意图是识别C个服从于子类型的样本聚类。
我们设每一个样本xi拥有一个标签指示向量(label indicator vector):
当xi属于第k个聚类时,
 ;否则,
。
;否则,
。
于是我们有划分矩阵(paitition matrix):
3.2 聚类方法
我们使用谱聚类算法(spectral clustering)得到网络划分:
其中
 为规模划分矩阵;
为规模划分矩阵;
 表示给定的相似度矩阵W的标准化拉普拉斯矩阵;矩阵D是网络的度的矩阵,对角线元素为对 应位置节点的度,非对角线元素设置为0。
表示给定的相似度矩阵W的标准化拉普拉斯矩阵;矩阵D是网络的度的矩阵,对角线元素为对 应位置节点的度,非对角线元素设置为0。
四、基于网络的生存风险预测
4.1 生存风险预测
给定所有的特征矩阵X,对于给定的第i为患者来说,一个事件(死亡)在 t 时间的风险为:
其中,z是回归系数向量;h0(t) 是基准风险函数。
而回归系数向量 z 通过最大限度的Cox logpartial似然估计出的:
其中n是患者的数量,ti 是第 i 位患者的生存时间;R(ti)是在ti 时间的风险集,表示的是在 ti 时间之前仍存活的患者集合;δi( )是一个指示器函数,当生存时间被观察到,δi = 1;否则为0。
4.2 改善的生存风险预测
我们可以通过附加信息改善生存预测,比如基因交互信息或基于约束的患者相似度。无论是功能的相似性还是患者的相似性(或两者)都可以进行正则化。
根据Cox模型的风险函数,患者i与患者j的相关风险为:
因此,可以创建一个正则项为:
为了预测z ,我们可以使用一个修改的函数:
其中λ是正则化系数。
基因组规模上的聚合数据类型的相似性网络融合摘要: 近期的技术已经使收集不同类型的全基因组数据十分划算,结合这些数据去创建一个给定的疾病或生物过程的一个全面视图的计算方法是有必要的。相似网络融合(SNF:similarity network fusion)通过创建每一个可利用的数据类型的样本(如,患者)的网络可以解决这个问题。例如,创建一个给定一群患者疾病的全面视图,SNF计算
相似性
网络
融合
是最初提出的一种技术,用于将来自不同来源的数据合并为一组共享的样本。
该过程的工作原理是为每个数据源构造这些样本的
网络
,以表示每个样本与所有其他样本的相似程度,然后将
网络
融合
在一起。
来自原始论文的此图将方法应用于遗传数据,提供了很好的演示:
相似性
网络
的生成和
融合
过程使用一种过程来降低样本之间较弱的关系的权重。
但是,在整个数据源之间保持一致的弱关系将通过
融合
过程得以保留。
有关
SNF
背后的数学
论文标题:
Similarity
network
fus
ion
for
agg
re
ga
ting
data
types
on a
genomic
scale
.
论文下载地址
论文以
计算
机视觉多视图方式为启发,设计了一种图
融合
网络
用于解决基因数据不能综合处理的困难。现有的基因数据非常丰富,有各种类型的基因数据可以利用。但现有的基因数据处理方式大多数是只利用一种基因数据,例如只使用DNA或者是只使用mRNA,不能综合所有的基因数据,得到一个既有共享信息又有互补信息的处理结果。本文考虑将患同一种癌症的
简单地说,缓存
融合
就是把Oracle RAC数据库中所有数据库缓存作为一个共享的数据库缓存,并被RAC中的所有节点共享。它是实现RAC的基本技术。
缓存
融合
主要有如下四个功能:
(1) 提供扩展性的传输。
(2) 在实例间传输数据库的映射。
(3) 跟踪资源的当前位置和状态。
(4) 在每个实例的S
GA
的目录结构中保存资源信息。
当把聚类(Clustering)和分类(Classificat
ion
)放到一起时,很容易弄混淆两者的概念,下分别对两个概念进行解释。
1 聚类(Clustering):
将物理或抽象对象的集合分成由类似的对象组成的多个类的过程被称为聚类。
聚类分析的一般做法是,先确定聚类统计量,然后利用统计量对样品或者变量进行聚类。对N个样品进行聚类的方法称为Q型

