wget方式下载(网络等问题可自行下载后上传服务器):
wget https://lammps.sandia.gov/tars/lammps-stable.tar.gz --no-check-certificate
tar -xzf lammps-stable.tar.gz
编译环境加载:
- Intel开发套件(parallel_studio_xe_2018)加载,如我组服务器为:
source /opt/intel/parallel_studio_xe_2018/psxevars.sh
- 高版本GCC加载(如v_7.5.0,此处采用module方式加载),因为makefile是由GNU make编写
module load GCC/7.5.0
进入lammps安装目录:
cd lammps-29Sep2021/src/
选择需要安装的package:
- 选择安装其它的包(如不需要,跳过此步)
如:make yes-kspace yes-manybody yes-molecule
或:make yes-all && make no-lib && make no-ext (!推荐,本教程采用!)
对于老版本: make yes-std && make no-lib
相关命令,详情见官方手册:
make yes-all # install all packages
make no-all # uninstall all packages
make yes-basic # install a few commonly used packages'
make no-basic # remove a few commonly used packages'
make yes-most # install most packages w/o libs'
make no-most # remove most packages w/o libs'
make yes-lib # install packages that require extra libraries
make no-lib # uninstall packages that require extra libraries
make yes-ext # install packages that require external libraries
make no-ext # uninstall packages that require external libraries
make package-status # show which packages are currently installed
make ps # show which packages are currently installed
make package-installed # show which packages are currently installed
make pi # show which packages are currently installed
注意:有些软件包需要其它库依赖,无法安装,详情见官方手册
Voronoi package的安装
采用官网手册中的Traditional make方法
make lib-voronoi # print help message
make lib-voronoi args="-b" # download and build the default version in lib/voronoi/voro++-<version>
make lib-voronoi args="-p $HOME/voro++" # use existing Voro++ installation in $HOME/voro++
make lib-voronoi args="-b -v voro++0.4.6" # download and build the 0.4.6 version in lib/voronoi/voro++-0.4.6
注意:下载voro+±0.4.6可能会网络出错导致lib/voronoi/Install.py脚本运行报错,此时自行下载并上传服务器并修改lib/voronoi/Install.py脚本。
voro++下载网站: http://math.lbl.gov/voro++/download/
voro+±0.4.6下载网址: http://math.lbl.gov/voro++/download/dir/voro++-0.4.6.tar.gz
- 下载完将安装包上传至
lammps-29Sep2021/lib/voronoi目录下 - 注释
Install.py脚本中的79行geturl(url, vorotar)
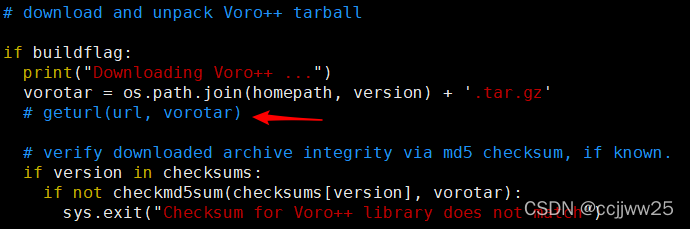
- 进入
lammps-29Sep2021/src/目录
make lib-voronoi args="-b -v voro++0.4.6"
make yes-voronoi
Colvars package的安装
采用官网手册中的Traditional make方法
make lib-colvars args="-m mpi"
make yes-colvars
开始编译(src/目录下):
make -j 12 intel_cpu_intelmpi
其中的12代表用12核cpu并行编译,若之前编译过或编译失败过,请在正式编译前运行make clean
linux系统下make方式安装lammps时,需要使用“make yes-包名称”的方式选择需要的包,如“make yes-MANYBODY"。在这个目录里,有个Install.py文件,运行这个文件会自动下载并编译安装Voro++库文件,前提是系统内已经装好python。但voronoi包不同于常规包,必须在make命令之前先进行下载和编译。
COLLECTIVE VARIABLES MODULE: Reference manual for LAMMPS
(‘collective variable’ shortened to colvar)
This manual documents the collective variables module (colvars), a portable software that interfaces
multiple MD simulation simulation programs, with a focus on flexibility, robustness and high performance.
本文介绍如何使用lammps命令计算单原子的体积。
lammps提供了compute voronoi/atom命令计算单原子体积。
基本原理可参考voronoi算法,根据voronoi算法,把单个原子所占据的空间划分为一个多边形,也称为泰森多边形,多边形的体积即为该原子的体积。
命令例句:
compute 1 all voronoi/atom
该例句共输出两个计算结果,第一个结果为单原子的体积,第二个结果为该原子多面体的面数,也可以理解为相邻原子的数目。
因此,引用单原子体积时,直
1.LAMMPS简介 LAMMPS由美国Sandia国家实验室开发,以GPL license发布,即开放源代码且可以免费获取使用,这意味着使用者可以根据自己的需要自行修改源代码。LAMMPS可以支持包括气态,液态或者固态相形态下、各种系综下、百万级的原子分子体系,并提供支持多种势函数,并且LAMMPS有良好的并行扩展性,适合大型HPC集群下的并行运行。2.安装环境需求1、硬件环...
运行你安装目录下的intel64/bin/mpivars.sh脚本(配置mpi environment)
打开host文件:cat ./hosts
并向host文件中写入所有mpi节点名称
下面是intel提供的一个例子:$ cat ./hosts
# this line is ignored
clusternode1

