


综述-药物发现中的分子设计:深度生成模型的全面回顾


今天为大家带来的是Yu Cheng等人于2021年11月于Briefings in Bioinformatics发表的文章 药物发现中的分子设计:深度生成模型的全面回顾。 下面由小编为大家带来解析。
在人类与疾病的长期斗争中,特别是最近2019年冠状病毒(COVID-19)而引起的大流感,药物正发挥着越来越重要的作用。然而, 药物研发过程 一直面临着许多障碍,需要大量的人力、物力和财力。 药物发现的困难在于化学分子的巨大和离散的搜索空间 。具体来说,类药化合物的可能结构规模在1023到1060之间,但只有其中一小部分是有治疗意义的。传统的方法,如高通量筛选是低效的,因为所需的资源量很大但是命中的机率却很小。大数据和高性能计算能力使人工智能超越了传统方法。随着深度学习的广泛应用,它自然被认为是药物发现的潜在途径。深度学习在药物发现和开发中的运用,为制药科学提供了一个新的方向。一些相关的应用如图1所示。
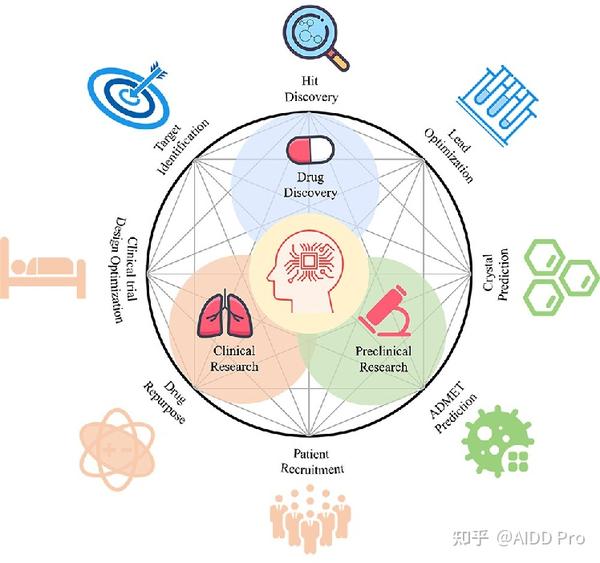

在这篇评论中,我们主要关注 药物发现中分子生成的深度生成模型 。我们首先介绍了分子的表示方法,和目前广为使用的数据库。我们展示了不同表示方法的优点和缺点。至于生成模型,我们强调了最近在新分子设计领域基于不同表示方法的进展。
分子生成中的分子表征和数据集
分子表征
在过去的几十年里,人们对计算方法产生了浓厚的兴趣,特别是随着 深度学习 的出现。这些技术的出现为 计算机辅助药物设计 打开了大门。这里的主要挑战是如何通过计算机准确地识别和存储分子,并为化学家所接受。在过去的几年里,由于计算机的快速发展,人们设计了大量的分子表征法。这里我们介绍两种在新分子设计中常用的表征方法,包括 SMILES表征 和 图表征 。
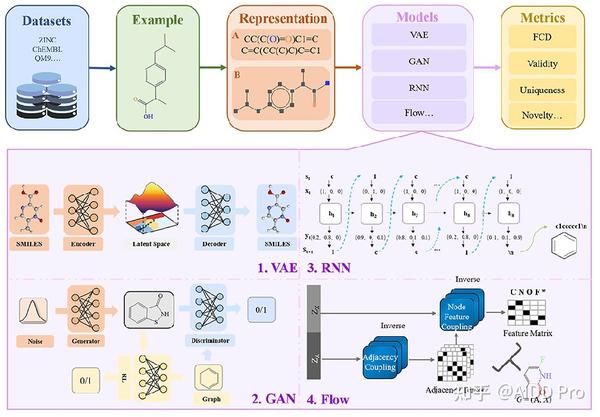
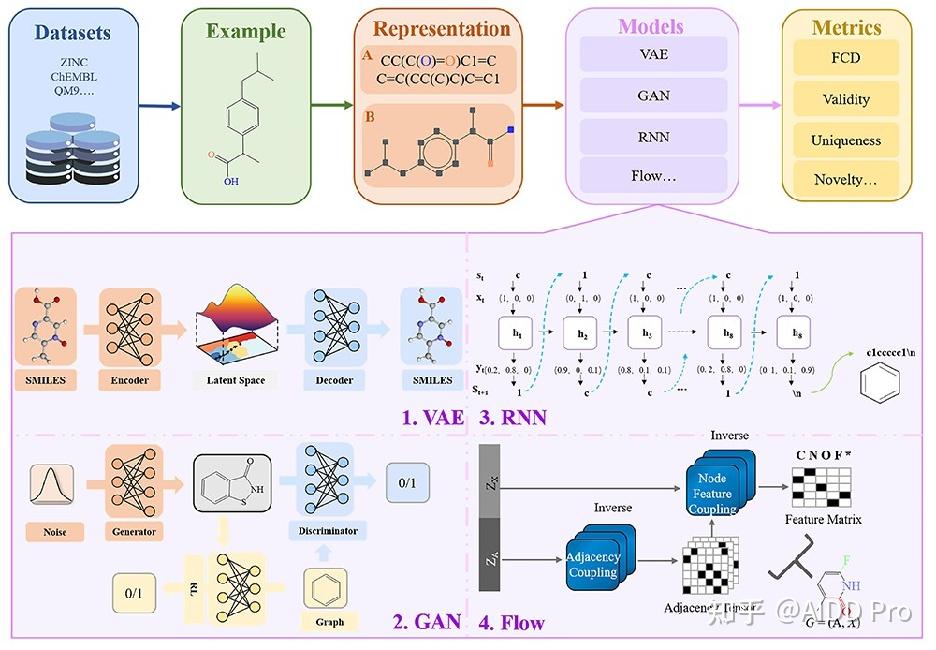
SMILES表征
SMILES表征主要使用线性字符串来表达化合物 ,它们很容易被计算机系统记忆和处理。一维线性表示法目前包括SMILES和国际化学标识符(InCHI)。 SMILES是一个ASCII字符串,它使用了从分子图到文本的映射算法,其中用严格的语法简化了化学结构 。一个分子的SMILES形式的例子见图2A。从分子结构到文本的转换使得SMILES易于被计算机处理,方便化学家使用,并易于用于训练机器学习模型。自动分子设计的第一个策略是使用基于SMILES的深度生成模型,并将这种表示方法转换为单热向量。但是SMILES也有一些缺点:(1)SMILES不能捕捉分子结构的相似性。两个相似结构之间的微小变化可能会导致SMILES字符串的巨大差异。2)SMILES字符串是非唯一的,一个分子可以被编码成多个SMILES表示。这些问题在目前的工作中已经多多少少地得到了解决。针对生成模型中典型的SMILES的不足,有许多研究产生了SMILES符号的变体并改进了模型。
图表征
SMILES是由基于图形的分子表示法产生的。而结构公式在化学中经常被用来表示分子。因此,描绘分子结构的一个更直观的方式是 分子图 。分子图形式的例子如图2B所示。每个分子可以表示为一个无向图G,其中节点集V和边集E分别由原子vi,和键(vi,vj)组成。具体来说,每个原子类型(碳、氢、氧.等)可以编码为T维的一热向量xi,而键的类型(单键、双键、三键和芳烃键)可以表示为y∈{1,...,Y}。随着图上深度学习研究的热潮,基于分子图的深度生成模型的训练在短短几年内兴起,目前已经有了许多应用。
数据集
机器学习中模型的训练是以数据为基础的,因此我们在这里重点讨论从头开始的分子设计中涉及的数据集。具体来说,我们将典型的分子生成模型中涉及的数据集分为以下几类。
第一类是 综合数据库 ,通常包含生物活性、化学结构和物理性质等多种信息,其中ZINC、ChEMBL、PubChem和DrugBank出现的频率较高。DrugBank中的药物数据字段可以与PubChem等其他数据库相连接。第二种是 合并的数据库 。这些数据库是通过合并和筛选现有的数据库而形成的化学数据集,不仅用于生成分子,而且还用于验证各种机器学习方法。
深度分子生成模型
近年来, 从头开始的分子设计 (de novo molecular design)是一个从头开始生成所需分子的概念,可以由专业专家或机器实现。由于 生成模型 的发展,与人类相比,分子生成不仅减少了化学分子的搜索空间,也减少了药物发现的时间消耗。在此,我们在下文中概述了一些基于两种经典表示法的典型分子生成模型,并在图3中总结了它们的时间轴。
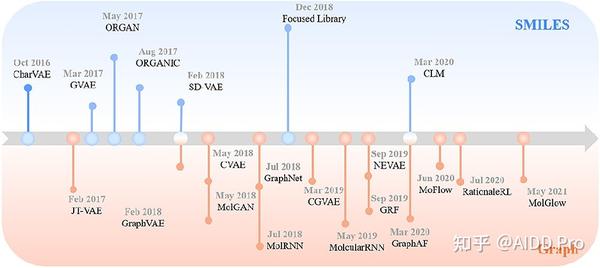

基于SMILES的模型
强大的深度学习技术推动了生成模型的发展。在对现实数据进行训练后,生成模型能够产生与给定样本相似的新合成数据。 深度生成模型 中有待解决的一个核心问题是如何捕捉未知的数据分布,并揭示内部的隐藏结构。其中一个方法是学习可以轻松建模的数据表征。在从头开始的分子设计领域,一个好的表征也是能够随时转换回分子的。就SMILES的简单特点而言,它已经被证明更容易被深度学习。而基于序列的方法可以进一步分为基于 变异编码器 、 生成对抗网络 (GANs)和 循环神经网 (RNNs)的模型。
基于图的模型
基于图的深度分子生成模型 一直是图研究中的一个热点,在药物发现方面有着广阔的前景。近三年来,在分子图生成领域有许多成功。考虑到SMILES上的VAE模型的成功,后来又开发了基于VAE的分子图设计的架构。Gómez-Bombarelli等人认为应进一步探索基于图的表示方法。此外,随着图神经网络的普及,基于图的模型也在新的分子设计中起到了主导作用。
结论
在这篇综述中,作者尽最大努力报告了分子生成演化路径的不同阶段,并强调了最近的研究进展。基于序列和基于图的生成模型都有其自身的优点。 分子生成模型的发展方式对药物发现起着重要的作用,它反映了深度神经网络在交叉领域的演变 。尽管已经取得了实质性的进展,但仍有很大的空间来提高现有生成模型的性能和改善合成可及性的指标。这些技术和计算能力的提升有望进一步提高生成具有良好设计的类药物特性的分子的质量,并进一步努力以完全自动化的方式加速新药设计。而这些分子生成的进展也预示着相关问题如逆向合成的美好前景。随着友好和易于使用的自动化工具的发展,化学家和计算机技术人员的合作工作将在未来进一步促进药物发现。
参考资料:
Yu Cheng, Yongshun Gong, Yuansheng Liu, Bosheng Song, Quan Zou, Molecular design in drug discovery: a comprehensive review of deep generative models, Briefings in Bioinformatics , Volume 22, Issue 6, November 2021, bbab344, https:// doi.org/10.1093/bib/bba b344
版权信息
本文系AIDD Pro接受的外部投稿,文中所述观点仅代表作者本人观点,不代表AIDD Pro平台,如您发现发布内容有任何版权侵扰或者其他信息错误解读,请及时联系AIDD Pro (请添加微信号sixiali_fox59)进行删改处理。
原创内容未经授权,禁止转载至其他平台。有问题可发邮件至sixiali@stonewise.cn