


BUSCO 评估基因组组装质量和完整性工具使用

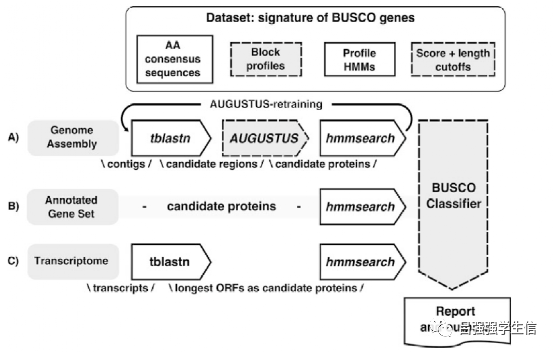
BUSCO是Benchmarking Universal Single-Copy Orthologs(通用单拷贝同源基因基准)的缩写,基于基因进化(有参比对)评估基因组组装和注释完整性的开源python软件。其对接结果的评估与 quast 不同,它并不追求基因组拼接的长度,而关注的是是否将一些单拷贝直系同源基因拼接出来。在相近的物种之间总有一些保守的序列,而 BUSCO 就是使用这些保守序列与组装的结果进行比对,鉴定组装的结果是否包含这些序列,包含单条、多条还是部分或者不包含等等情况来给出结果。
BUSCO 评估的原理:
软件根据 OrthoDB 数据库,构建了几个大的进化分支的单拷贝基因集。使用hmmsearch进行比对,将拼接结果预测得到的基因集与该基因集进行比较,根据比对上的比例、完整性,来评价拼接结果的准确性和完整性。也就是比对上已知基因集的基因越多,说明拼接的结果越好。
使用hmmsearch进行比对时:
根据得分判断单拷贝蛋白质是否存在:
expected score’ cut-off is defined as 90% of the minimum bitscore from an HMM search of all of a BUSCO group’s members against its own HMM profile
根据长度判断是否完整:
这里长度根据该单拷贝基因家族的长度分布,query的长度必须落在平均长度的±2σ 之间即为完整,当BUSCO中的蛋白在query蛋白集中比对到多个,则被认为是多拷贝。
一:软件安装:
下载链接:
https://gitlab.com/ezlab/busco/
https://busco.ezlab.org/
https://busco.ezlab.org/busco_userguide.html
其依赖比较多,还需要配置Python环境,建议用Conda安装。
Conda安装命令
conda create -n busco -c conda-forge -c bioconda busco=5.3.2下图是安装过程:
二.下载数据库文件
软件安装完毕之后,就开始下载数据库文件了,根据组装的物种来选择对应的数据库文件。以节肢动物数据库下载为例
https:// busco-data.ezlab.org/v5 /data/lineages/
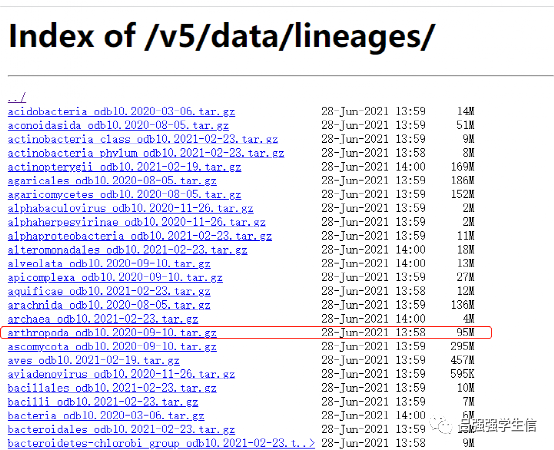
https:// busco-data.ezlab.org/v5 /data/lineages/arthropoda_odb10.2020-09-10.tar.gz
顺便下载了真核生物的、后生生物的、蛛形纲的。
https:// busco-data.ezlab.org/v5 /data/lineages/eukaryota_odb10.2020-09-10.tar.gz
https:// busco-data.ezlab.org/v5 /data/lineages/metazoa_odb10.2021-02-24.tar.gz
https:// busco-data.ezlab.org/v5 /data/lineages/arachnida_odb10.2020-08-05.tar.gz
tar -zxvf arthropoda_odb10.2020-09-10.tar.gz
cd arthropoda_odb10/
三.参数设置
公司给建议:
busco --config config.ini -i genome.fa -r -o sample_name --out_path ./ -l arthropoda_odb10 -m geno -c 32 -f
我的运行命令:
busco -i /path/to/canu_removedup.fa -r -o canu_remdup --out_path /path/to/busco --lineage_dataset /path/to/arthropoda_odb10 -m geno -c 32 -f --offline
这行命令依赖metaeuk寻找可能的编码区,还可以通过augustus进行:
busco -i /path/to/canu_removedup.fa -r -o canu_remdup_augus --out_path / path/to /busco --lineage_dataset / path/to /arthropoda_odb10 -m geno -c 32 -f --offline --augustus
--augustus: Use augustus gene predictor for eukaryote runs
-i or --in 指定输入文件,核苷酸序列或者蛋白的fasta序列文件,v5.1.0,输入文件可以指定为目录文件,对文件夹内的fasta进行批量处理;