-
报错如下图:

这个问题困扰了好久,看这个信息也没看到是哪里出错,然后一行一行执行。最终找到了
rf.confusionMatrix <- confusionMatrix(rf.class , data.test$Class, positive = "T")
这行代码是通过预测出来的结果和测试集比较,生成混淆矩阵。那么这个levels指的就是T或者F。也就是说可能有数据没匹配上或者变量类型不是factor。我们一步一步验证。
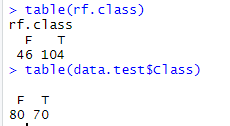
- 首先用table函数查看数据条目,可以看到,数据条目是一样的,那么就不是这个问题。
- 接下来查看变量类型,如下图所示,变量类型不一样。
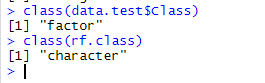
- 看到这里,我们就知道解决办法了,我们需要把预测结果转化为factor类型。
rf.confusionMatrix <- confusionMatrix(factor(rf.class) , data.test$Class, positive = "T")
StackOverflow-1
StackOverflow-2
谢谢点击进来,如果您觉得有用,麻烦点赞鼓励一下!报错如下图:这个问题困扰了好久,看这个信息也没看到是哪里出错,然后一行一行执行。最终找到了rf.confusionMatrix <- confusionMatrix(rf.class , data.test$Class, positive = "T")这行代码是通过预测出来的结果和测试集比较,生成混淆矩阵。那么这个levels指的就...
数据读写对离散变量,我们会观测变量各个层级观测的频数,或者使用两个变量的交叉表格,对离散变量绘制条形图等;
对连续变量,我们会看某个变量的均值,标准差,分位数等
此外,summary(),str(),describe(()等函数(psych包里)做义工数据框的总结。
以上即为一些最基础的方法,但这些方法灵活性不高,输出的信息也是固定的,这时我们需要对数据进行整形。
在整合和整形操作前,我们
下载数据(人类)
由于我需要获取剪切位点两边的序列,那么我需要下载参考基因组数据和注释文件。参考基因组下载常用的有ncbi、ucsc和ensemble。下图是参考基因组版本对应信息。
我是从NCBI下载
链接(https://www.ncbi.nlm.nih.gov/)
点击搜索之后,就可以在页面中找到了。
GenCode
我的注释文件是在GenCode下载,下面为版本信息
这个错误通常是由于在进行模型训练或预测时,数据集中的某些变量的因子水平不一致导致的。因子(Factor)是R中的一种数据类型,它表示分类变量,并将其存储为整数,每个整数对应于一个类别。在进行模型训练或预测时,如果训练集和测试集中的某些变量的因子水平不一致,则会导致上述错误。
解决这个错误的方法是确保数据集中的所有因子变量的因子水平一致。具体来说,可以使用 `factor()` 函数将因子变量转换为因子类型,并使用 `levels()` 函数来查看因子变量的因子水平。
示例代码:
# 假设数据集为data,其中x和y是因子变量
data$x <- factor(data$x)
data$y <- factor(data$y)
# 检查因子变量x和y的因子水平
levels(data$x)
levels(data$y)
# 假设测试集为test_data,其中x和y是因子变量
test_data$x <- factor(test_data$x)
test_data$y <- factor(test_data$y)
# 检查因子变量x和y的因子水平
levels(test_data$x)
levels(test_data$y)
如果发现因子变量的因子水平不一致,则可以使用 `factor()` 函数和 `levels()` 函数来调整数据集中的因子变量,使得其因子水平一致。

