


常用生信SCI图绘制代码(R语言)

本篇文章将生物学中常用的图形代码,整理到一块。
示例数据都是基于网络的, 代码可以直接复制运行 。注释齐全。
另外如果不想写代码,在每个图形后面还附有一个 在线绘图 的网址。
内容持续更新中!
1. 热图
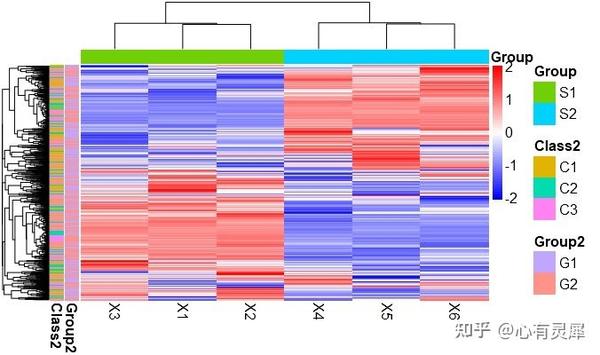
library(pheatmap) # 加载pheatmap这个R包
# 1,读取热图数据文件
df = read.delim("https://www.bioladder.cn/shiny/zyp/demoData/heatmap/data.heatmap.txt", #文件名称 注意文件路径,格式
header = T, # 是否有标题
sep = "\t", # 分隔符是Tab键
row.names = 1, # 指定第一列是行名
fill=T) # 是否自动填充,一般选择是
# (可选)读取分组数据文件
# dfSample = read.delim("https://www.bioladder.cn/shiny/zyp/demoData/heatmap/sample.class.txt",header = T,row.names = 1,fill = T,sep = "\t")
# dfGene = read.delim("https://www.bioladder.cn/shiny/zyp/demoData/heatmap/gene.class.txt",header = T,row.names = 1,fill = T,sep = "\t")
# 2,绘图
pheatmap(df,
# annotation_row=dfGene, # (可选)指定行分组文件
# annotation_col=dfSample, # (可选)指定列分组文件
show_colnames = TRUE, # 是否显示列名
show_rownames=TRUE, # 是否显示行名
fontsize=2, # 字体大小
color = colorRampPalette(c('#0000ff','#ffffff','#ff0000'))(50), # 指定热图的颜色
annotation_legend=TRUE, # 是否显示图例
border_color=NA, # 边框颜色 NA表示没有
scale="row", # 指定归一化的方式。"row"按行归一化,"column"按列归一化,"none"不处理
cluster_rows = TRUE, # 是否对行聚类
cluster_cols = TRUE # 是否对列聚类
)详细的内容可参考:
不想写代码?可以用BioLadder生信云平台在线绘图:
2. PCA
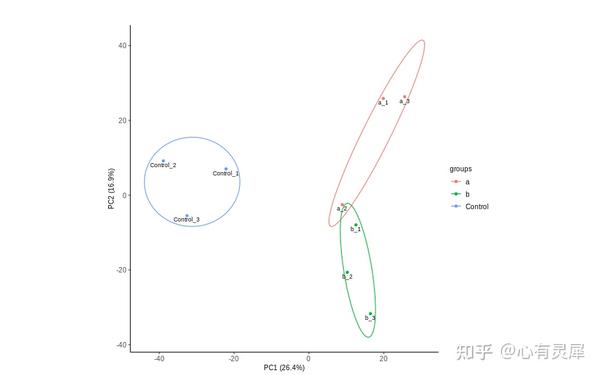
# 加载R包,没有安装请先安装 install.packages("包名")
library(ggplot2)
library(ggbiplot)
# ggbiplot包需要从github上下载
# install.packages("devtools")
# library(devtools)
# install_github("vqv/ggbiplot")
# 读取PCA数据文件
df = read.delim("https://www.bioladder.cn/shiny/zyp/bioladder2/demoData/PCA/data.txt",# 这里读取了网络上的demo数据,将此处换成你自己电脑里的文件
header = T, # 指定第一行是列名
row.names = 1 # 指定第一列是行名
df=t(df) # 对数据进行转置,如果想对基因分组则不用转置
# 读取样本分组数据文件
dfGroup = read.delim("https://www.bioladder.cn/shiny/zyp/bioladder2/demoData/PCA/sample.class.txt",
header = T,
row.names = 1
# PCA计算
pca_result <- prcomp(df,
scale=T # 一个逻辑值,指示在进行分析之前是否应该将变量缩放到具有单位方差
ggbiplot(pca_result,
var.axes=F, # 是否为变量画箭头
obs.scale = 1, # 横纵比例
groups = dfGroup[,1], # 添加分组信息,为分组文件的第一列
ellipse = T, # 是否围绕分组画椭圆
circle = F)+
geom_text( # geom_text一个在图中添加标注的函数
aes(label=rownames(df)), # 指定标注的内容为数据框df的行名
vjust=1.5, # 指定标记的位置,vjust=1.5 垂直向下1.5个距离。 负数为位置向上标记,正数为位置向下标记
size=2 # 标记大小
theme_bw()
详细的内容可以参考:
不想写代码?可以用BioLadder生信云平台在线绘图:
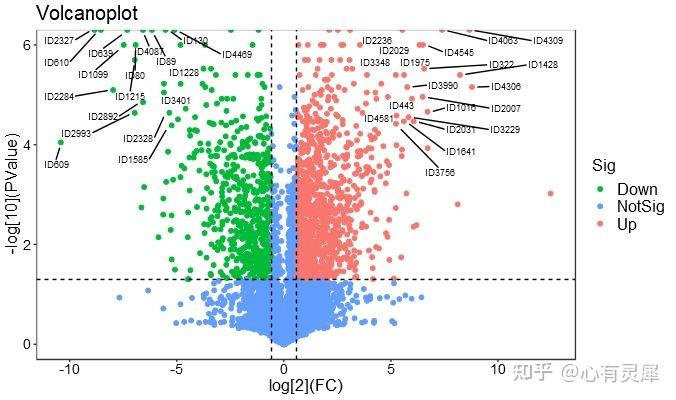
3. 火山图

# 加载R包,没有安装请先安装 install.packages("包名")
library(ggplot2)
library(ggrepel) #用于标记的包
# 读取火山图数据文件
data = read.delim("https://www.bioladder.cn/shiny/zyp/bioladder2/demoData/Volcano/Volcano.txt",# 这里读取了网络上的demo数据,将此处换成你自己电脑里的文件
header = T # 指定第一行是列名
# 建议您的文件里对应的名称跟demo数据一致,这样不用更改后续代码中的变量名称
FC = 1.5 # 用来判断上下调,一般蛋白质组的项目卡1.5
PValue = 0.05 #用来判断上下调
# 判断每个基因的上下调,往数据框data里新增了sig列
data$sig[(-1*log10(data$PValue) < -1*log10(PValue)|data$PValue=="NA")|(log2(data$FC) < log2(FC))& log2(data$FC) > -log2(FC)] <- "NotSig"
data$sig[-1*log10(data$PValue) >= -1*log10(PValue) & log2(data$FC) >= log2(FC)] <- "Up"
data$sig[-1*log10(data$PValue) >= -1*log10(PValue) & log2(data$FC) <= -log2(FC)] <- "Down"
# 标记方式(一)
# 根据数据框中的Marker列,1的为标记,0的为不标记
data$label=ifelse(data$Marker == 1, as.character(data$Name), '')
# (或)标记方式(二)
# 根据PValue小于多少和log[2]FC的绝对值大于多少筛选出合适的点
# PvalueLimit = 0.0001
# FCLimit = 5
# data$label=ifelse(data$PValue < PvalueLimit & abs(log2(data$FC)) >= FCLimit, as.character(data$Name), '')
ggplot(data,aes(log2(data$FC),-1*log10(data$PValue))) + # 加载数据,定义横纵坐标
geom_point(aes(color = sig)) + # 绘制散点图,分组依据是数据框的sig列
labs(title="volcanoplot", # 定义标题,x轴,y轴名称
x="log[2](FC)",
y="-log[10](PValue)") +
# scale_color_manual(values = c("red","green","blue")) + # 自定义颜色,将values更改成你想要的三个颜色
geom_hline(yintercept=-log10(PValue),linetype=2)+ # 在图上添加虚线
geom_vline(xintercept=c(-log2(FC),log2(FC)),linetype=2)+ # 在图上添加虚线
geom_text_repel(aes(x = log2(data$FC), # geom_text_repel 标记函数
y = -1*log10(data$PValue),
label=label),
max.overlaps = 10000, # 最大覆盖率,当点很多时,有些标记会被覆盖,调大该值则不被覆盖,反之。
size=3, # 字体大小
box.padding=unit(0.5,'lines'), # 标记的边距
point.padding=unit(0.1, 'lines'),
segment.color='black', # 标记线条的颜色
show.legend=FALSE)+
theme_bw()详细的内容可以参考:
不想写代码?可以用BioLadder生信云平台在线绘图:
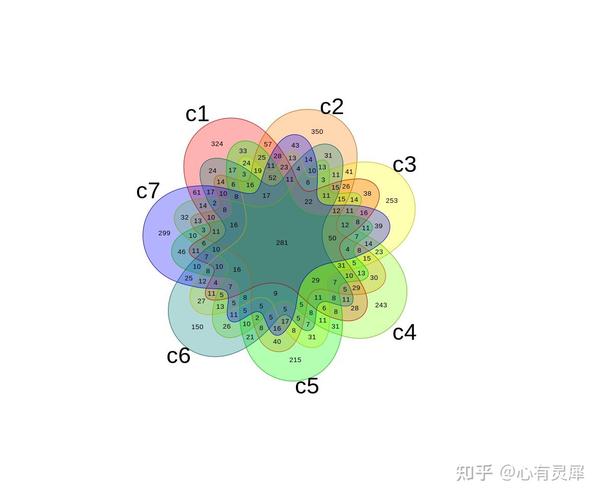
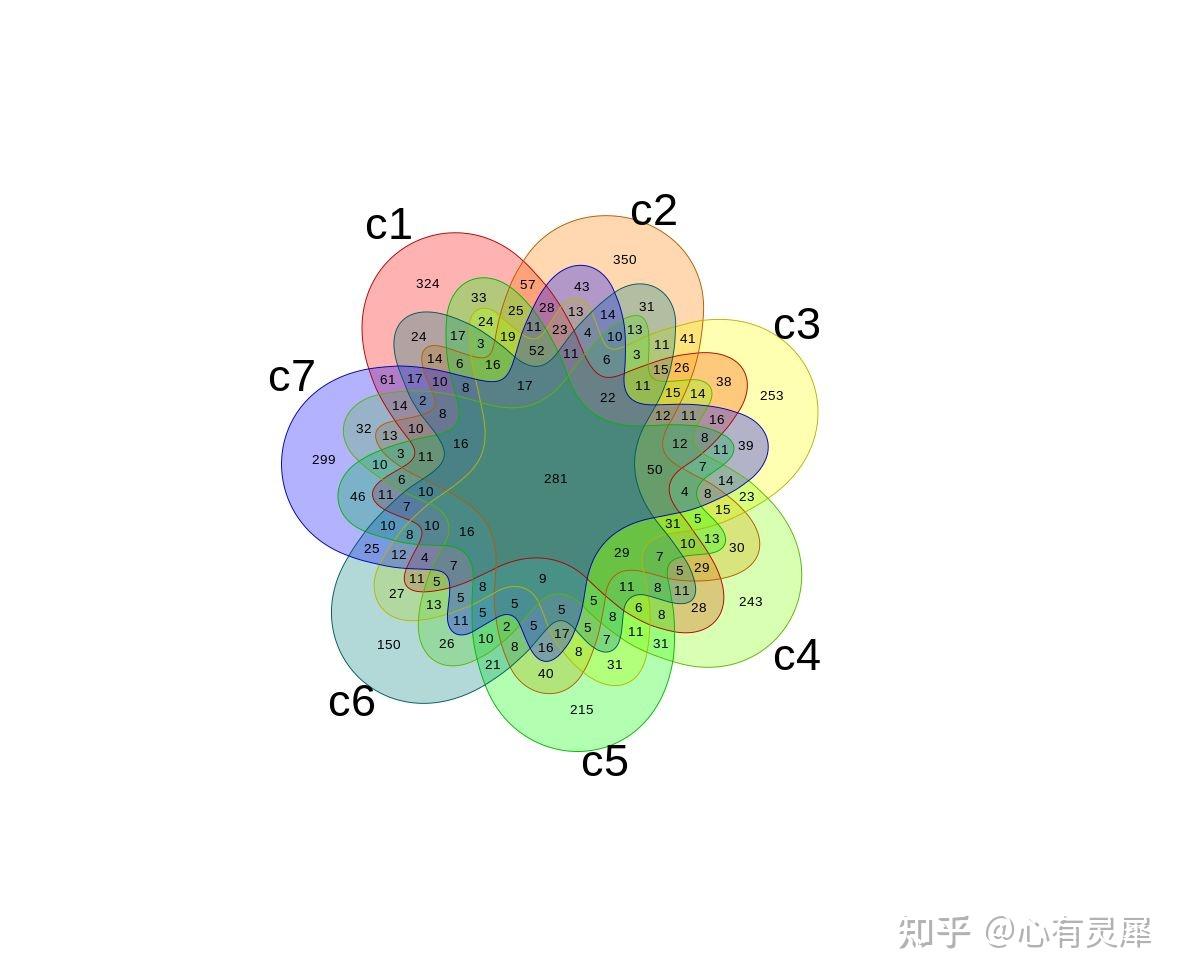
4. 韦恩图
# 加载R包,没有安装请先安装 install.packages("包名")
library(venn) #韦恩图(venn 包,适用样本数 2-7)
library(VennDiagram)
# 读取数据文件
venn_dat <- read.delim('https://www.bioladder.cn/shiny/zyp/bioladder2/demoData/Venn/flower.txt') # 这里读取了网络上的demo数据,将此处换成你自己电脑里的文件
venn_list <- list(venn_dat[,1], venn_dat[,2], venn_dat[,3], venn_dat[,4], venn_dat[,5], venn_dat[,6], venn_dat[,7]) # 制作韦恩图搜所需要的列表文件
names(venn_list) <- colnames(venn_dat[1:7]) # 把列名赋值给列表的key值
venn(venn_list,
zcolor='style', # 调整颜色,style是默认颜色,bw是无颜色,当然也可以自定义颜色
opacity = 0.3, # 调整颜色透明度
box = F, # 是否添加边框
ilcs = 0.5, # 数字大小
sncs = 1 # 组名字体大小
# 更多参数 ?venn查看
# 查看交集详情,并导出结果
inter <- get.venn.partitions(venn_list)
for (i in 1:nrow(inter)) inter[i,'values'] <- paste(inter[[i,'..values..']], collapse = '|')
inter <- subset(inter, select = -..values.. )
inter <- subset(inter, select = -..set.. )
write.table(inter, "result.csv", row.names = FALSE, sep = ',', quote = FALSE)详细的内容可以参考:
不想写代码?可以用BioLadder生信云平台在线绘图:
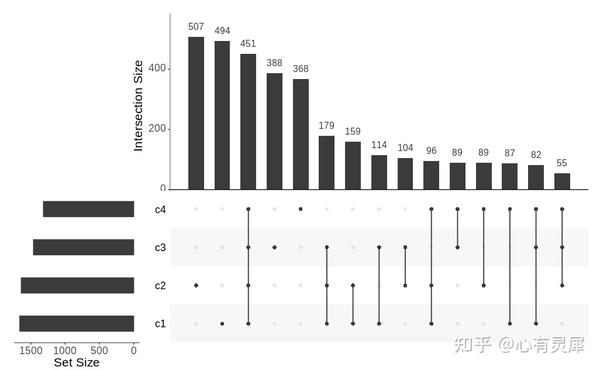
5. Upest图
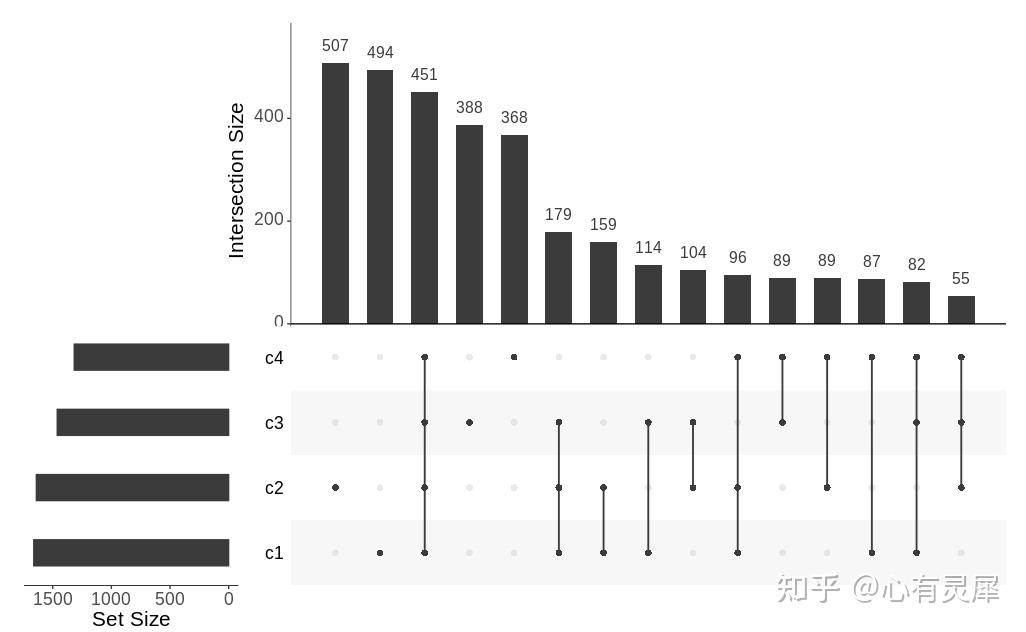
# 加载R包,没有安装请先安装 install.packages("包名")
library(UpSetR) #Upset图(upset 包,适用样本数 2-7)
library(VennDiagram)
# 读取数据文件
upset_dat <- read.delim('https://www.bioladder.cn/shiny/zyp/bioladder2/demoData/Venn/flower.txt') # 这里读取了网络上的demo数据,将此处换成你自己电脑里的文件
upset_list <- list(upset_dat[,1], upset_dat[,2], upset_dat[,3], upset_dat[,4], upset_dat[,5], upset_dat[,6], upset_dat[,7]) # 制作Upset图搜所需要的列表文件
names(upset_list) <- colnames(upset_dat[1:7]) # 把列名赋值给列表的key值
upset(fromList(upset_list), # fromList一个函数,用于将列表转换为与UpSetR兼容的数据形式。
nsets = 100, # 绘制的最大集合个数
nintersects = 40, #绘制的最大交集个数,NA则全部绘制
order.by = "freq", # 矩阵中的交点是如何排列的。 "freq"根据交集个数排序,"degree"根据
keep.order = F, # 保持设置与使用sets参数输入的顺序一致。默认值是FALSE,它根据集合的大小排序。
mb.ratio = c(0.6,0.4), # 左侧和上方条形图的比例关系
text.scale = 2 # 文字标签的大小
# 更多参数 ?upset查看
# 查看交集详情,并导出结果
inter <- get.venn.partitions(upset_list)
for (i in 1:nrow(inter)) inter[i,'values'] <- paste(inter[[i,'..values..']], collapse = '|')
inter <- subset(inter, select = -..values.. )
inter <- subset(inter, select = -..set.. )
write.table(inter, "result.csv", row.names = FALSE, sep = ',', quote = FALSE)详细的内容可参考:
不想写代码?可以用BioLadder生信云平台在线绘图:
6. 3维PCA
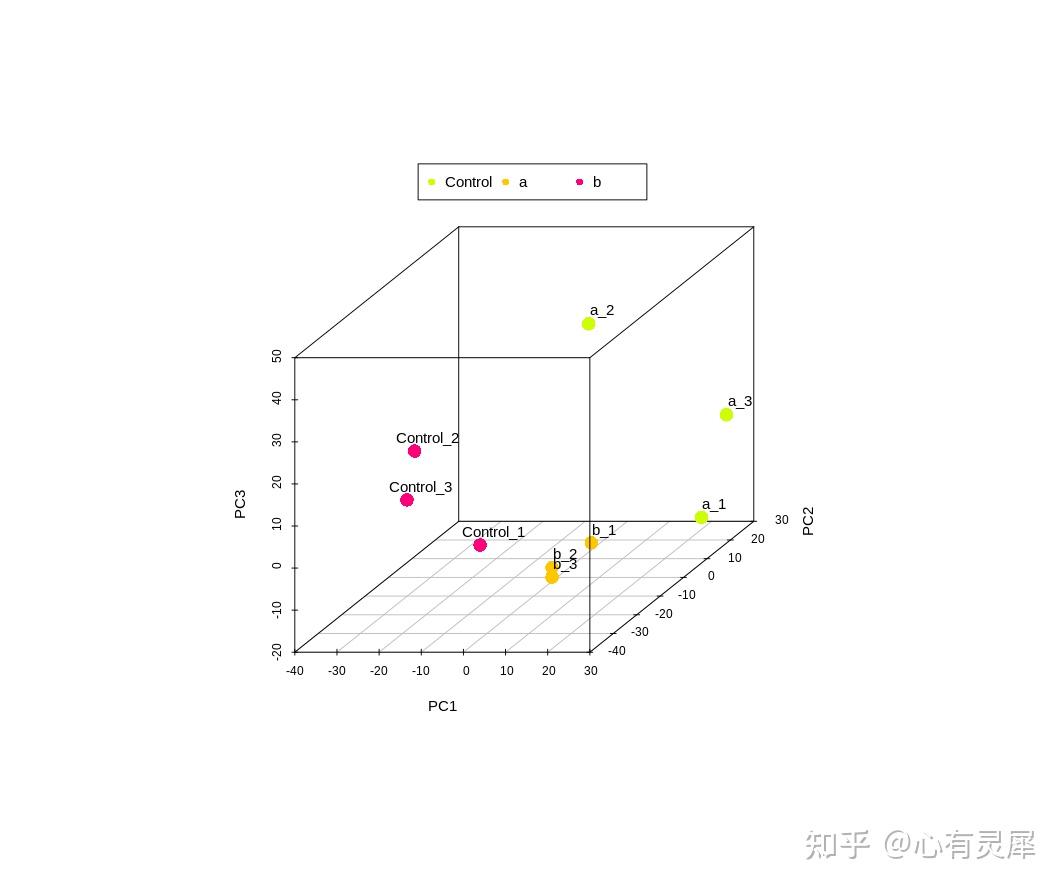
# 加载R包,没有安装请先安装 install.packages("包名")
library(scatterplot3d)
# 读取PCA数据文件
df = read.delim("https://www.bioladder.cn/shiny/zyp/bioladder2/demoData/PCA/data.txt",# 这里读取了网络上的demo数据,将此处换成你自己电脑里的文件
header = T, # 指定第一行是列名
row.names = 1 # 指定第一列是行名
df=t(df) # 对数据进行转置,如果想对基因分组则不用转置
# 读取样本分组数据文件
dfGroup = read.delim("https://www.bioladder.cn/shiny/zyp/bioladder2/demoData/PCA/sample.class.txt",
header = T,
row.names = 1
# PCA计算
pca_result <- prcomp(df,
scale=T # 一个逻辑值,指示在进行分析之前是否应该将变量缩放到具有单位方差
pca_result$x<-data.frame(pca_result$x)
# 设置颜色,有几个分组就写几个颜色
colors <- c("red","blue","green")
colors <- colors[as.numeric(as.factor(dfGroup[,1]))]
# 设置点形状,仅供参考
# shape<-16:18
# shape<-shape[as.numeric(as.factor(dfGroup[,1]))]
# 计算PC值,并替换列名,用来替换坐标轴上的标签
# pVar <- pca_result$sdev^2/sum(pca_result$sdev^2)
# pVar = round(pVar,digits = 3)
# colnames(pca_result$x) = c(
# paste0("PC1 (",as.character(pVar[1] * 100 ),"%)"),
# paste0("PC2 (",as.character(pVar[2] * 100 ),"%)"),
# paste0("PC3 (",as.character(pVar[3] * 100 ),"%)"),
# "PC4",
# "PC5",
# "PC6",
# "PC7",
# "PC8",
# "PC9"
s3d <- scatterplot3d(pca_result$x[,1:3],
pch = 16, # 点形状
color=colors, # 点颜色
cex.symbols = 2 # 点大小
# 设置图例
legend("top",
legend = unique(dfGroup[,1]),
col = c("red","blue","green"),
pch = 16,
inset = -0.1,
xpd = TRUE,
horiz = TRUE)
# 设置文字标注
text(s3d$xyz.convert(pca_result$x[,c(1,2,3)] + 2),
labels = row.names(pca_result$x),
cex = 0.8,col = "black")详细的内容可以参考:
不想写代码?可以用BioLadder生信云平台在线绘图:
7. ROC曲线

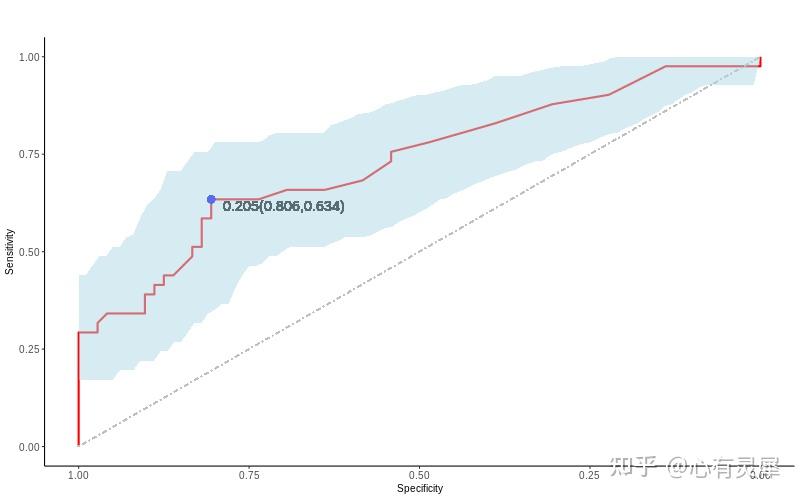
# 加载R包,没有安装请先安装 install.packages("包名")
library(pROC)
library(ggplot2)
# 读取ROC数据文件
df = read.delim("https://www.bioladder.cn/shiny/zyp/bioladder2/demoData/ROC/demo.txt",# 这里读取了网络上的demo数据,将此处换成你自己电脑里的文件
header = T # 指定第一行是列名
# ROC计算
rocobj <- roc(df[,1], df[,2],
# controls=df[,2][df[,1]=="Good"], # 可以设置实验组或对照组
# cases=df[,2][df[,1]=="Poor"],
smooth = F # 曲线是否光滑,当光滑时,无法计算置信区间
# 计算临界点/阈值
cutOffPoint <- coords(rocobj, "best")
cutOffPointText <- paste0(round(cutOffPoint[1],3),"(",round(cutOffPoint[2],3),",",round(cutOffPoint[3],3),")")
# 计算AUC值
auc<-auc(rocobj)[1]
# AUC的置信区间
auc_low<-ci(rocobj,of="auc")[1]
auc_high<-ci(rocobj,of="auc")[3]
# 计算置信区间
ciobj <- ci.se(rocobj,specificities=seq(0, 1, 0.01))
data_ci<-ciobj[1:101,1:3]
data_ci<-as.data.frame(data_ci)
x=as.numeric(rownames(data_ci))
data_ci<-data.frame(x,data_ci)
ggroc(rocobj,
color="red",
size=1,
legacy.axes = F # FALSE时 横坐标为1-0 specificity;TRUE时 横坐标为0-1 1-specificity
theme_classic()+
geom_segment(aes(x = 1, y = 0, xend = 0, yend = 1), # 绘制对角线
colour='grey',
linetype = 'dotdash'
geom_ribbon(data = data_ci, # 绘制置信区间
aes(x=x,ymin=X2.5.,ymax=X97.5.), # 当legacy.axes=TRUE时, 把x=x改为x=1-x
fill = 'lightblue',
alpha=0.5)+
geom_point(aes(x = cutOffPoint[[2]],y = cutOffPoint[[3]]))+ # 绘制临界点/阈值
geom_text(aes(x = cutOffPoint[[2]],y = cutOffPoint[[3]],label=cutOffPointText),vjust=-1) # 添加临界点/阈值文字标签详细的内容可参考:
不想写代码?可以用BioLadder生信云平台在线绘图:
8. 雷达图
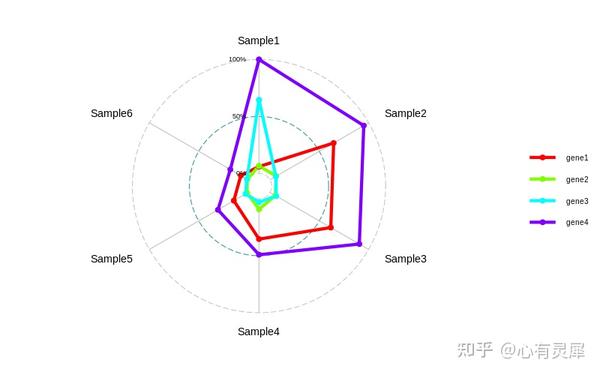
# 加载R包,没有安装请先安装 install.packages("包名")
library(ggradar) # 此包需要从github安装 devtools::install_github("ricardo-bion/ggradar")
library(ggplot2)
# 读取雷达图数据文件
df = read.delim("https://www.bioladder.cn/shiny/zyp/bioladder2/demoData/radarChart/demo.csv",# 这里读取了网络上的demo数据,将此处换成你自己电脑里的文件
sep = ",", # 指定文件分隔符
header = T # 指定第一行是列名
ggradar(df,
grid.max = max(df[,-1]), # 设置坐标轴的最大值
grid.mid = max(df[,-1])/2, # 设置坐标轴的中间值
grid.min = 0, # 设置坐标轴的最小值
grid.label.size = 4, # 坐标轴百分比标签大小
axis.label.size = 5, # 组名标签字体大小
group.colours = rainbow(length(df[,1])), # 设置颜色,数量跟第一列的个数相同
background.circle.colour = "white", # 设置背景颜色
group.point.size = 2, # 点大小
group.line.width = 2, # 线条粗细
plot.legend = T, # 是否显示图例
legend.position = "right", # 图例位置"top", "right", "bottom", "left"
legend.title = "", # 图例标题
legend.text.size = 10, # 图例文字大小
plot.title = "Title", # 标题名称
plot.extent.x.sf = 1.2, # 设置图片横向延伸空间,防止外圈文字显示不全
plot.extent.y.sf = 1.2, # 设置图片纵向延伸空间,防止外圈文字显示不全
# 更多选项?ggradar查看,支持ggplot2其他函数扩展详细的内容可参考:
不想写代码?可以用BioLadder生信云平台在线绘图:
9. 箱线图
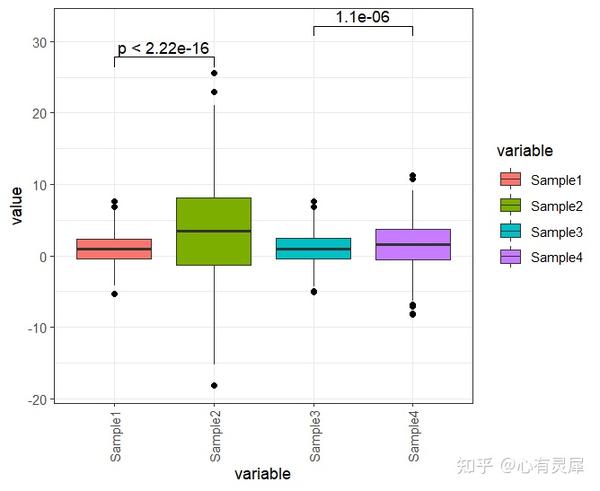
# 加载R包,没有安装请先安装 install.packages("包名")
library(ggplot2)
library(reshape2) # 用于转换数据
library(ggsignif) # 用于添加显著性标签
# 读取箱线图数据文件
df = read.delim("https://www.bioladder.cn/shiny/zyp/bioladder2/demoData/BoxPlot/boxplot.txt",# 这里读取了网络上的demo数据,将此处换成你自己电脑里的文件
header = T # 指定第一行是列名
# 把数据转换成ggplot常用的类型(长数据)
df = melt(df)
ggplot(df,aes(x=variable,y=value,fill=variable))+
# stat_boxplot(geom = "errorbar", # 添加误差线
# width=0.3)+
geom_boxplot(alpha = 1, # 透明度
outlier.color = "black" # 外点颜色
theme_bw()+ # 白色主题
theme(
axis.text.x = element_text(angle = 90,
vjust = 0.5
) # x轴刻度改为倾斜90度,防止名称重叠
geom_signif( # 添加显著性标签
comparisons=list(c("Sample1","Sample2"),c("Sample3","Sample4")), # 选择你想在哪组上添加标签
step_increase = 0.1,
test="t.test", # "t 检验,比较两组(参数)" = "t.test","Wilcoxon 符号秩检验,比较两组(非参数)" = "wilcox.test"
map_signif_level=F # 标签样式F为数字,T为*号
)详细的内容可参考:
不想写代码?可以用BioLadder生信云平台在线绘图:
10. 小提琴图
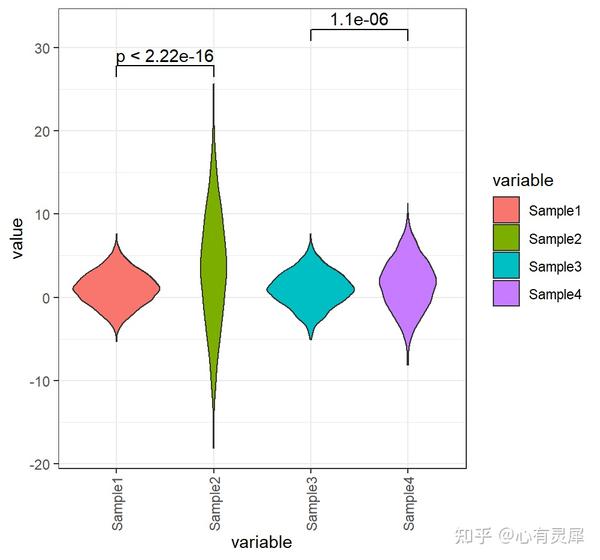
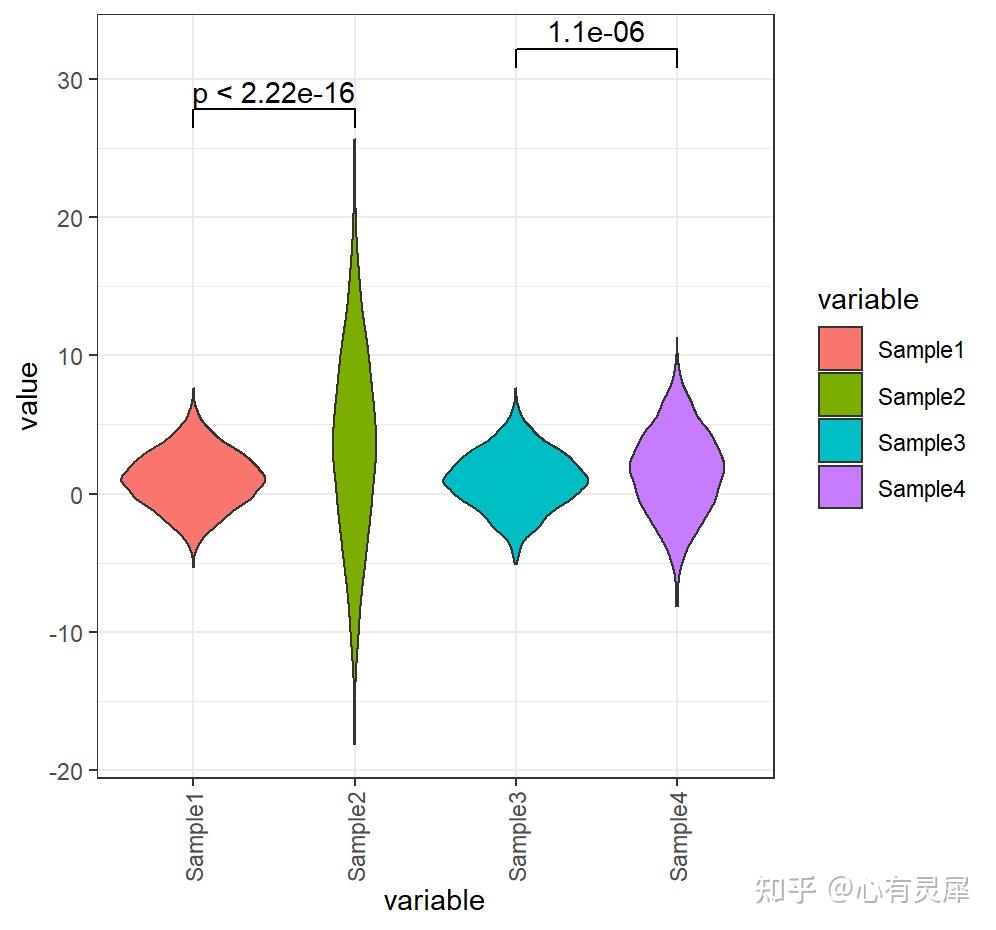
# 加载R包,没有安装请先安装 install.packages("包名")
library(ggplot2)
library(reshape2)
library(ggsignif)
# 读取小提琴图数据文件
df = read.delim("https://www.bioladder.cn/shiny/zyp/bioladder2/demoData/BoxPlot/boxplot.txt",# 这里读取了网络上的demo数据,将此处换成你自己电脑里的文件
header = T # 指定第一行是列名
# 把数据转换成ggplot常用的类型(长数据)
df = melt(df)
ggplot(df,aes(x=variable,y=value,fill=variable))+
geom_violin(alpha = 1, # 透明度
trim = T, # 是否修剪尾巴,即将数据控制到真实的数据范围内
scale = "area", # 如果“area”(默认),所有小提琴都有相同的面积(在修剪尾巴之前)。如果是“count”,区域与观测的数量成比例。如果是“width”,所有的小提琴都有相同的最大宽度。
theme_bw()+ # 白色主题
theme(
axis.text.x = element_text(angle = 90,
vjust = 0.5
) # x轴刻度改为倾斜90度,防止名称重叠
geom_signif( # 添加显著性标签
comparisons=list(c("Sample1","Sample2"),c("Sample3","Sample4")), # 选择你想在哪2组上添加标签
step_increase = 0.1,
test="t.test", # "t 检验,比较两组(参数)" = "t.test","Wilcoxon 符号秩检验,比较两组(非参数)" = "wilcox.test"
map_signif_level=F # 标签样式F为数字,T为*号
)详细的内容可参考:
不想写代码?可以用BioLadder生信云平台在线绘图:
11. 核密度图
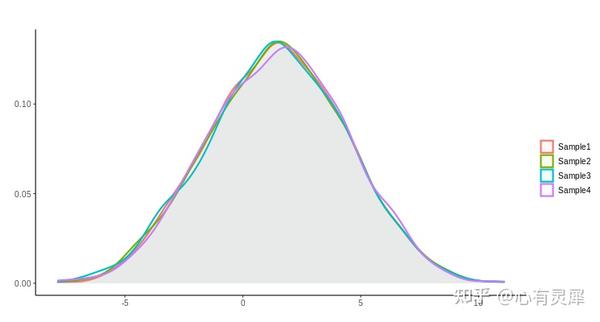
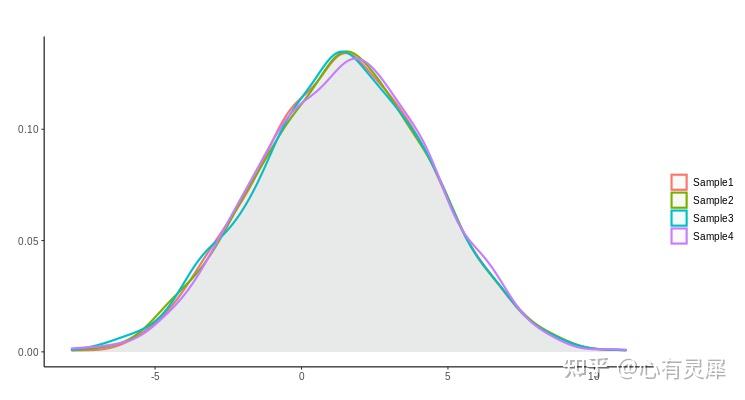
# 加载R包,没有安装请先安装 install.packages("包名")
library(ggplot2)
library(reshape2)
# 读取核密度图数据文件
df = read.delim("https://www.bioladder.cn/shiny/zyp/bioladder2/demoData/BoxPlot/boxplot.txt",# 这里读取了网络上的demo数据,将此处换成你自己电脑里的文件
header = T # 指定第一行是列名
# 把数据转换成ggplot常用的类型(长数据)
df = melt(df) # melt出自reshape2包
head(df) # 查看转换完成的数据的前几行
ggplot(df,aes(x=value,
fill=variable, # fill填充颜色,根据变量名赋值
colour=variable))+ # colour图形边界颜色,根据变量名赋值
geom_density(alpha=0.2, # 填充颜色透明度
size=1, # 线条粗细
linetype = 1 # 线条类型1是实线,2是虚线
theme_bw() # 白色主题
# 补充知识:
# fill 一般是指填充颜色
# color 一般是指线和点的颜色
# colour 一般是指图形边界颜色详细的内容可以参考:
不想写代码?可以用BioLadder生信云平台在线绘图:
12. 双向柱形图

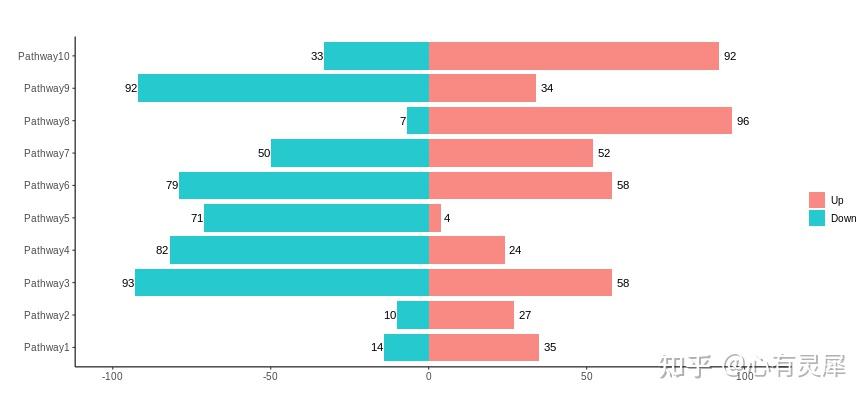
# 加载R包,没有安装请先安装 install.packages("包名")
library(ggplot2)
library(reshape2)
# 读取双向柱形图数据文件
df= read.delim("https://www.bioladder.cn/shiny/zyp/bioladder2/model/ggplot2/DoublePositionBarPlot/demo.txt") # 这里读取了网络上的demo数据,将此处换成你自己电脑里的文件
# 把数据转换成ggplot常用的类型(长数据)
df = melt(df) # melt出自reshape2包
head(df) # 查看转换完成的数据的前几行
# X variable value
# 1 Pathway1 Up 35
# 2 Pathway2 Up 27
# 3 Pathway3 Up 58
# 4 Pathway4 Up 24
# 5 Pathway5 Up 4
# 6 Pathway6 Up 58
ggplot(df, aes(
x = factor(X,levels = unique(X)), # 将第一列转化为因子,目的是显示顺序与文件顺序相同,否则按照字母顺序排序
y = ifelse(variable == "Up", value, -value), # 判断分组情况,将两个柱子画在0的两侧
fill = variable)) +
geom_bar(stat = 'identity')+ # 画柱形图
coord_flip()+ # x轴与y轴互换位置
geom_text( # 在图形上加上数字标签
aes(label=value, # 标签的值(数据框的第三列)
# vjust = ifelse(variable == "Up", -0.5, 1), # 垂直位置。如果没有coord_flip(),则可以取消这行注释
hjust = ifelse(variable == "Up", -0.4, 1.1) # 水平位置
size=2 # 标签大小
scale_y_continuous( # 调整y轴
labels = abs, # 刻度设置为绝对值
expand = expansion(mult = c(0.1, 0.1))) # 在y轴的两侧,留下一部分的空白位置,防止加标签的时候,显示不全详细的内容可以参考:
不想写代码?可以用BioLadder生信云平台在线绘图:
13. 相关性热图
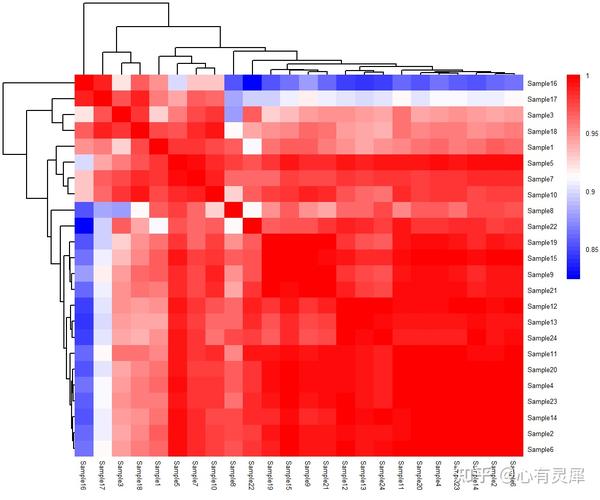

# 加载R包,没有安装请先安装 install.packages("包名")
library(pheatmap)
# 读取相关性热图数据文件
df= read.delim("https://www.bioladder.cn/shiny/zyp/bioladder2/demoData/Cor/demo.txt",# 这里读取了网络上的demo数据,将此处换成你自己电脑里的文件
header = T,
row.names = 1
# 计算相关性
r <- cor(df,
method = "pearson", # 计算相关性的方法有"pearson", "spearman", "kendall"
use = "pairwise.complete.obs" # 缺失值处理的方式
# 绘制热图
pheatmap(r,
show_colnames = TRUE, # 是否显示列名
show_rownames=TRUE, # 是否显示行名
fontsize=5, # 字体大小
color = colorRampPalette(c('#0000ff','#ffffff','#ff0000'))(50), # 指定热图的颜色
annotation_legend=TRUE, # 是否显示图例
border_color=NA, # 边框颜色 NA表示没有
scale="none", # 指定归一化的方式。"row"按行归一化,"column"按列归一化,"none"不处理
cluster_rows = TRUE, # 是否对行聚类
cluster_cols = TRUE # 是否对列聚类
)详细的内容可以参考:
不想写代码?可以用BioLadder生信云平台在线绘图:
14. 相关性矩阵
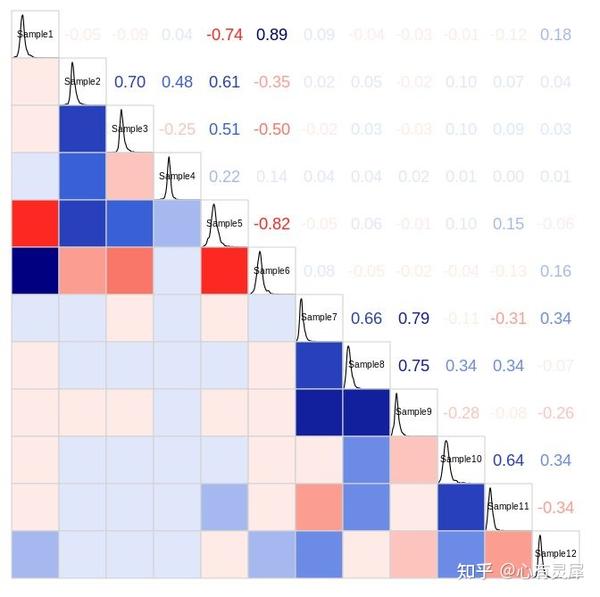

# 加载R包,没有安装请先安装 install.packages("包名")
library(corrgram)
# 读取相关性矩阵数据文件
df= read.delim("https://www.bioladder.cn/shiny/zyp/bioladder2/demoData/Cor/demo2.txt",# 这里读取了网络上的demo数据,将此处换成你自己电脑里的文件
header = T,
row.names = 1
corrgram(df,
cex.labels = 0.8, # 样本文字大小
lower.panel="panel.pts", # 指定下方面板的形状,具体参数看下方注释
upper.panel="panel.cor", # 指定上方面板的形状,具体参数看下方注释
diag.panel="panel.density",# 指定中间面板的形状,具体参数看下方注释
cor.method="pearson") # 计算相关性的方法有"pearson", "spearman", "kendall"
# 上、下面板可供选择的图形
# "散点" = "panel.pts"
# "柱形" = "panel.bar"
# "相关系数(带置信区间)"="panel.conf"
# "相关系数(带颜色)"="panel.cor"
# "ellipse"="panel.ellipse"
# "填充"="panel.fill"
# "饼图"="panel.pie"
# "阴影"="panel.shade"
# "空"=NULL
# 中间面板可供选择的图形
# "核密度图" = "panel.density"
# "最大最小值" = "panel.minmax"
# "空"=NULL
# 更多参数可以输入 ?corrgram 查看详细的内容可参考:
不想写代码?可以用BioLadder生信云平台在线绘图:
14. 富集气泡图
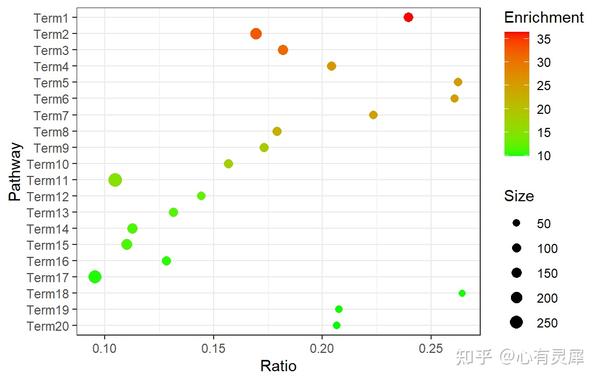
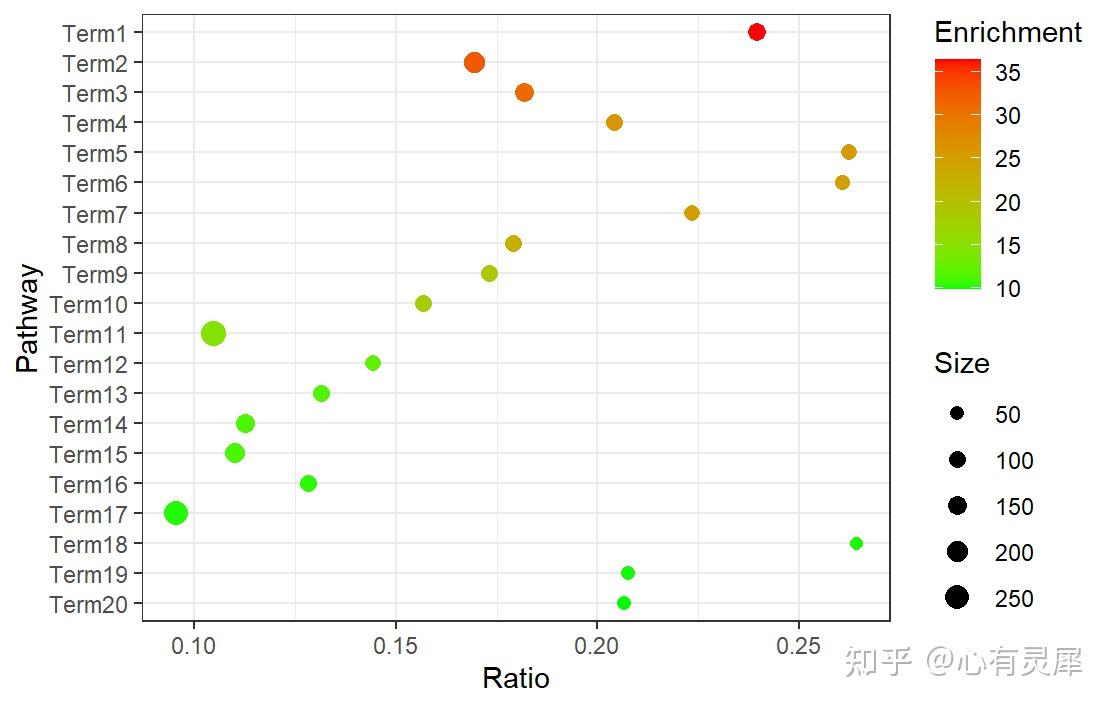
library(ggplot2)
# 读取富集气泡图数据文件
df= read.delim("https://www.bioladder.cn/shiny/zyp/bioladder2/demoData/bubble/data.txt")# 这里读取了网络上的demo数据,将此处换成你自己电脑里的文件
ggplot(df,aes(x = Ratio,
y = reorder(Term,Enrichment,sum), # 按照富集度大小排序
size = Size,
colour=Enrichment)) +
geom_point(shape = 16) + # 设置点的形状
labs(x = "Ratio", y = "Pathway")+ # 设置x,y轴的名称
scale_colour_continuous( # 设置颜色图例
name="Enrichment", # 图例名称
low="green", # 设置颜色范围
high="red")+
scale_radius( # 设置点大小图例
range=c(2,4), # 设置点大小的范围
name="Size")+ # 图例名称
guides(
color = guide_colorbar(order = 1), # 决定图例的位置顺序
size = guide_legend(order = 2)
theme_bw() # 设置主题详细的内容可参考:
不想写代码?可以用BioLadder生信云平台在线绘图:
15. 词云图


library(jiebaRD)
library(jiebaR)
library(wordcloud2)
# 先将一段文字转成词频
text = "蛋白质组(Proteome)的概念最先由Marc Wilkins提出,指由一个基因组(genome),或一个细胞、组织表达的所有蛋白质(Protein). 蛋白质组的概念与基因组的概念有许多差别,它随着组织、甚至环境状态的不同而改变. 在转录时,一个基因可以多种mRNA形式剪接,并且,同一蛋白可能以许多形式进行翻译后的修饰. 故一个蛋白质组不是一个基因组的直接产物,蛋白质组中蛋白质的数目有时可以超过基因组的数目. 蛋白质组学(Proteomics)处于早期“发育”状态,这个领域的专家否认它是单纯的方法学,就像基因组学一样,不是一个封闭的、概念化的稳定的知识体系,而是一个领域. 蛋白质组学集中于动态描述基因调节,对基因表达的蛋白质水平进行定量的测定,鉴定疾病、药物对生命过程的影响,以及解释基因表达调控的机制. 作为一门科学,蛋白质组研究并非从零开始,它是已有20多年历史的蛋白质(多肽)谱和基因产物图谱技术的一种延伸. 多肽图谱依靠双向电泳(Two-dimensional gel electrophoresis, 2-DE)和进一步的图象分析;而基因产物图谱依靠多种分离后的分析,如质谱技术、氨基酸组分分析等."
initialize = worker(stop_word="stopwords.txt")
# 停止词文件可以在这里下载https://www.bioladder.cn/shiny/zyp/bioladder2/demoData/wordcloud/stopwords.txt
df<-freq(initialize[text]) # 计算词频
wordcloud2(df,
size = 1, # 字体大小
fontFamily = 'Segoe UI', # 字体
fontWeight = 'bold', # 字体粗细
color = 'random-dark', # 字体颜色
backgroundColor = "white", # 背景颜色
minRotation = -pi/4, # minRotation和maxRotation控制文本旋转角度的范围
maxRotation = pi/4,
rotateRatio = 0.4, # 文本旋转的概率 0.4表示大约有40%的词发生了旋转
shape = "circle" # 轮廓形状
)详细的内容可参考:
不想写代码?可以用BioLadder生信云平台在线绘图:
16. 多边形树状图
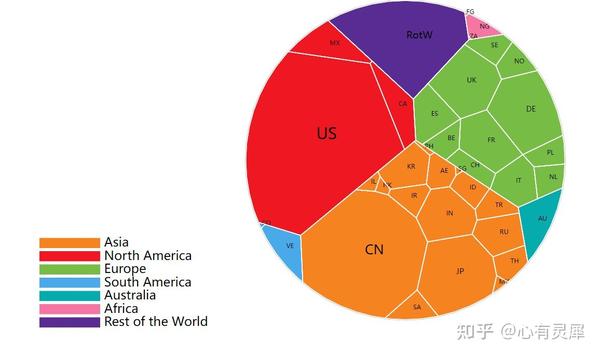
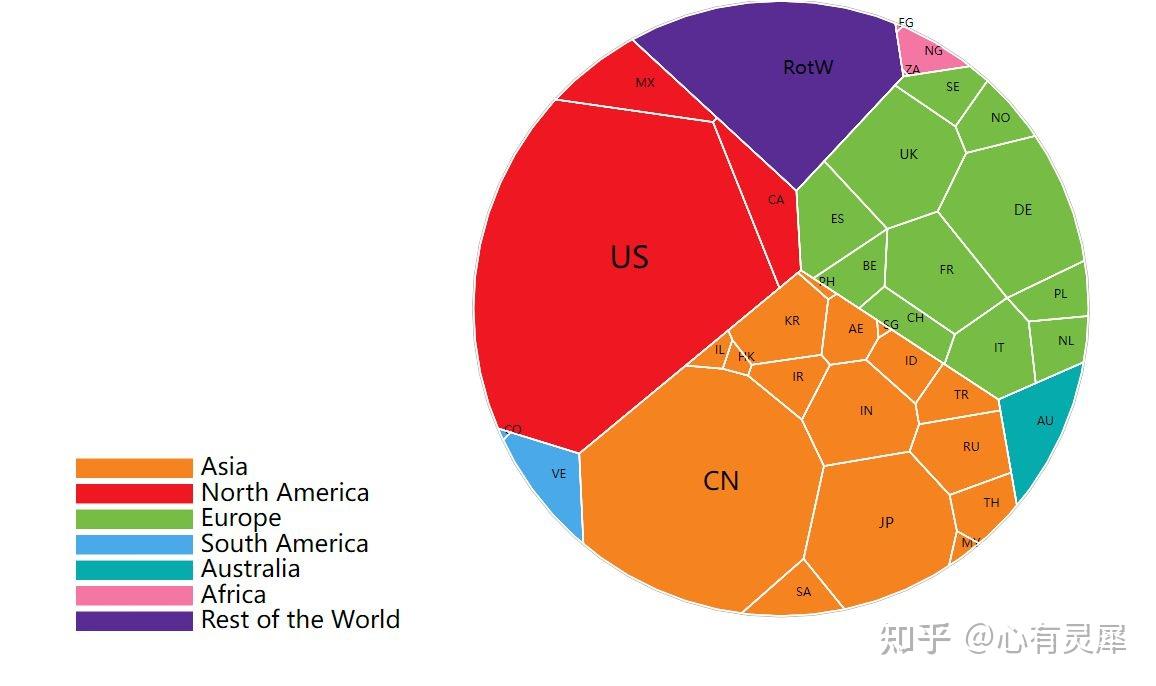
# voronoiTreemap包的安装方式为
# devtools::install_github("uRosConf/voronoiTreemap")
library(voronoiTreemap)
# 读取多边形树状图数据文件
df= read.delim("https://www.bioladder.cn/shiny/zyp/bioladder2/demoData/treemap/2.txt")# 这里读取了网络上的demo数据,将此处换成你自己电脑里的文件
# 生成层次结构
data_int = vt_input_from_df(df,
hierachyVar0 = "h1", # 定义哪列是第1个层次
hierachyVar1 = "h2", # 定义哪列是第2个层次
hierachyVar2 = "h3", # 定义哪列是第3个层次
colorVar = "color", # 定义每个多边形的颜色
weightVar = "size", # 定义每个多边形的大小
labelVar = "abbreviation"# 定义每个多边形的缩写标签
vt_d3(vt_export_json(data_int),
legend= T, # 是否显示图例
label = T # 是否显示文字标签
)详细的内容可参考:
不想写代码?可以用BioLadder生信云平台在线绘图:
17. 时间序列分析
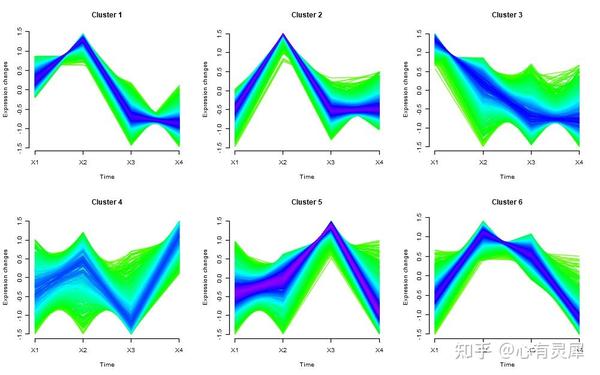
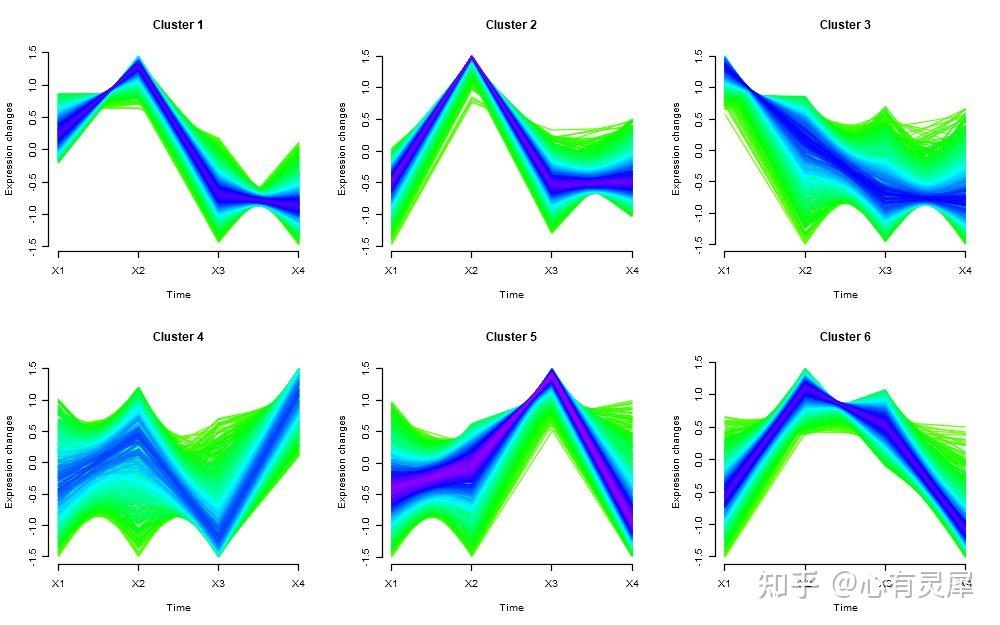
# Mfuzz包的安装方式为:BiocManager::install("Mfuzz")
library(Mfuzz)
# 读取时间序列分析数据文件
df = read.delim("https://www.bioladder.cn/shiny/zyp/bioladder2/demoData/mfuzz/all_gene_fpkm.xls",# 这里读取了网络上的demo数据,将此处换成你自己电脑里的文件
row.names = 1
# 构建对象,填补缺失值,标准化等
dm <- data.matrix(df) # 数据框转换为矩阵
ESet <- new("ExpressionSet",exprs = dm) # 构建对象
ESet <- filter.NA(ESet, thres=0.25) # 过滤缺失值超过“25%”的基因
ESet <- fill.NA(ESet,mode="knn") # knn算法填补缺失值
ESet <- filter.std(ESet,min.std=0) # 根据标准差去除样本间差异太小的基因
gene.s <- standardise(ESet) # 标准化
exprs(gene.s) # 查看处理后的数据
c <- 6 # 设置聚类个数
m <- mestimate(gene.s) # 评估出最佳的m值
set.seed(123) # 设置随机种子,防止每次聚类的结果都不一样,无法复现
cl <- mfuzz(gene.s, c = c, m = m)
cl # 查看每个基因聚到哪个类当中
cl$size # 查看每个cluster中的基因个数
cl$membership # 查看基因和cluster之间的membership。如果两个基因对于一个特定的cluster都有高的membership score,那么他们通常来说表达模式是相似的
mfuzz.plot(
gene.s,
mfrow=c(2,3),# 图形排列方式,2行3列
new.window= FALSE) # 是否在新窗口打开详细的内容可参考:
不想写代码?可以用BioLadder生信云平台在线绘图:
18. 云雨图
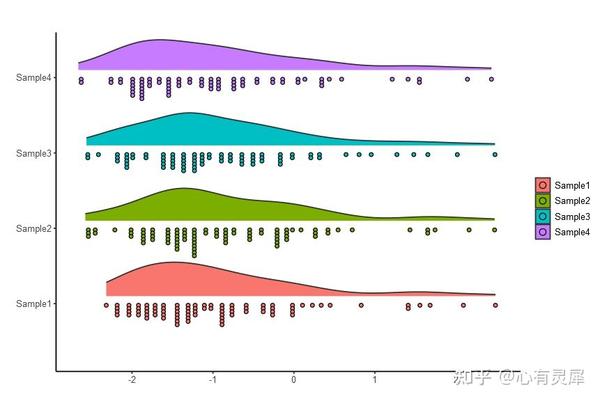
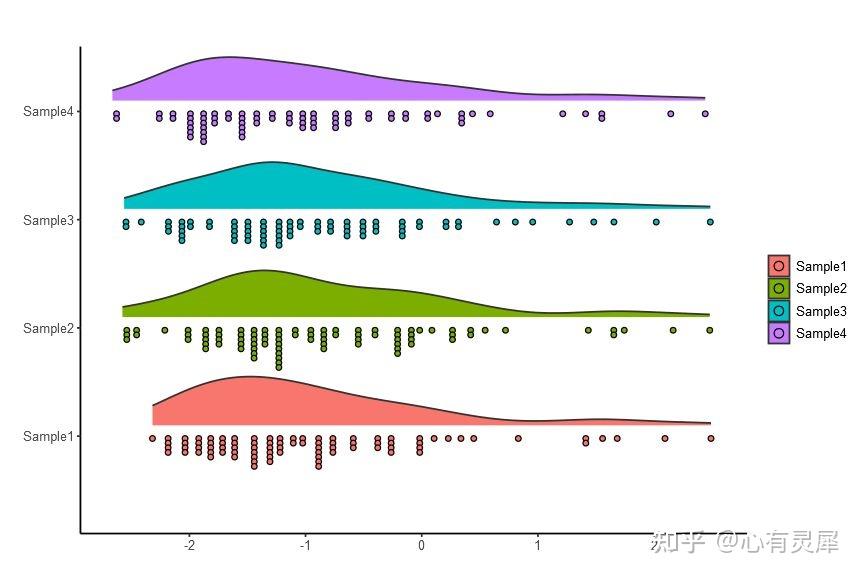
详细的内容可以参考:
不想写代码?可以用BioLadder生信云平台在线绘图:
19. 棒棒糖图
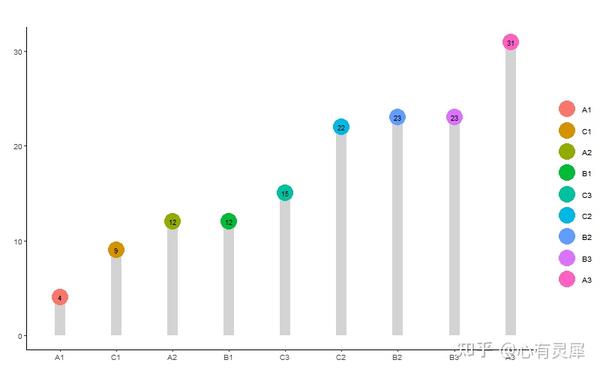
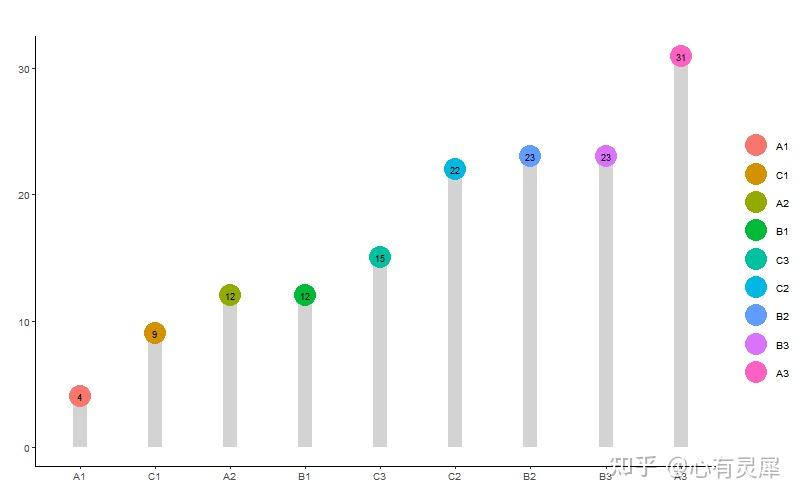
# 加载R包,没有安装请先安装 install.packages("包名")
library(ggpubr)
library(ggplot2)
# 读取棒棒糖图数据文件
df = read.delim("https://www.bioladder.cn/shiny/zyp/bioladder2/demoData/lolly/demoData.txt")
ggdotchart(df,
x = colnames(df)[1],
y = colnames(df)[2],
color = colnames(df)[1],
palette = rainbow(dim(df)[1]), # 修改颜色
sorting = "none", # 排列方式 sorting = c("ascending", "descending", "none"),
add = "segments", # 添加线条add = c("none", "segment")
dot.size = 10, # 点大小
add.params = list(color = "lightgray", size = 5), #修改线条参数,颜色,粗细
position = position_dodge(0.1), #调整位置
label = round(df[[2]]), #添加数字标签
font.label = list(color = "white", # 设置数字标签参数,颜色,大小,位置
size = 9,
vjust = 0.5)
)详细的内容可参考:
不想写代码?可以用BioLadder生信云平台在线绘图:
20. 累积曲线
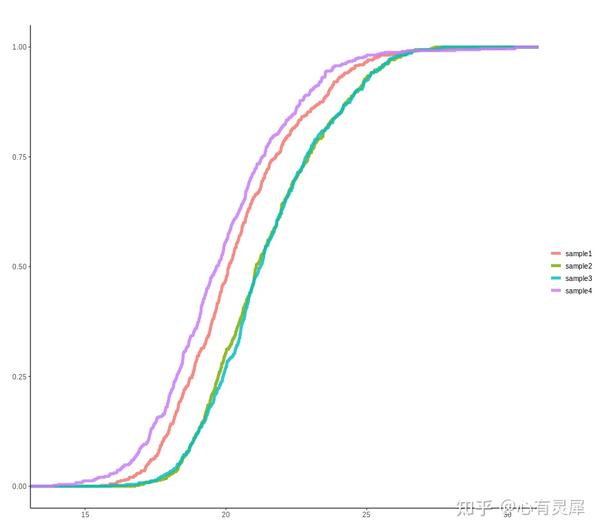
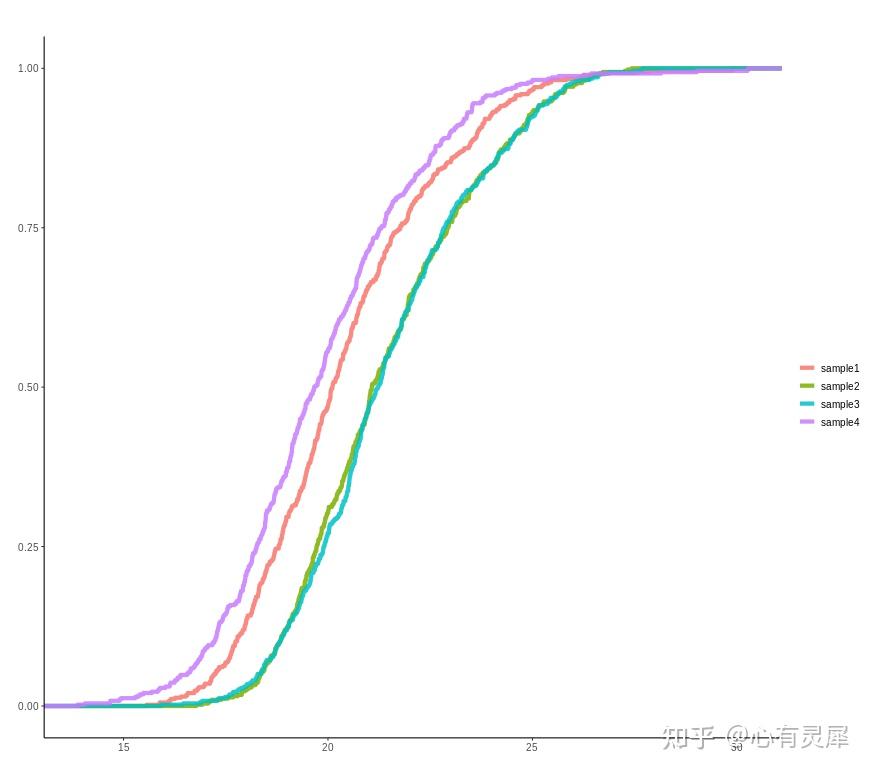
# 加载R包,没有安装请先安装 install.packages("包名")
library(ggplot2)
library(reshape2)
# 读取累积曲线数据文件
df = read.delim("https://www.bioladder.cn/shiny/zyp/bioladder2/demoData/ecdf/demo.txt",
row.names = 1)
df = melt(df) # reshape2中的函数,宽数据转换为长数据
df$value = log(df$value) # 为方便观察,做log变化,以实际数据是否需要为准
ggplot(df,
x=value,
color=variable
stat_ecdf( # ggplot2中的经验累积分布函数
size=2 # 线条粗细
)详细的内容可参考:
不想写代码?可以用BioLadder生信云平台在线绘图:
21. 频率分布直方图

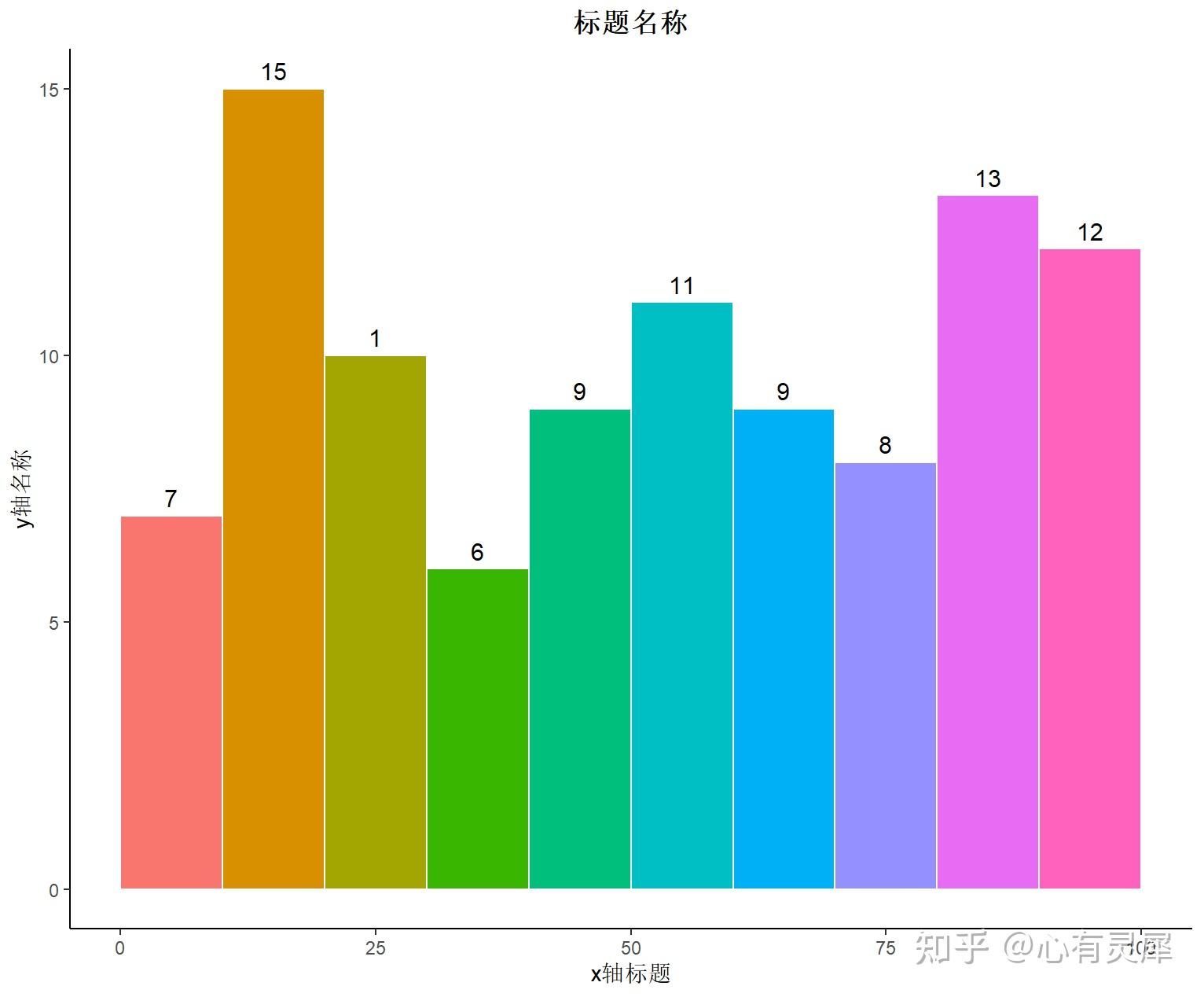
library(ggplot2)
# 创建数据框1到100的随机数100个
random = sample(1:100, 100, replace = T)
df = data.frame(random)
seq = seq(0,100,10) # 提前设置区间
ggplot(df,
aes(x=df[,1],
fill=cut(df[,1],seq) # 添加颜色,填充依据是seq
geom_histogram(breaks=seq, # 设置break区间
color="white", # 柱子的边框颜色
size=0.5, # 边框大小
stat="bin")+
geom_text( #加标签
aes(label=gsub("^0$", "", as.character(..count..))), # 内容为统计数字并去除0
stat="bin",
vjust=-0.5, # 垂直位置
size=4, # 标签大小
breaks=seq)+ # 标签位置
theme_classic()+
theme(
legend.position = "none", # 隐藏图例
plot.title = element_text( # 标题居中并加粗
face = "bold",
hjust = 0.5)
labs( # 调整坐标轴名称
x = "x轴标题",
y = "y轴名称",
title = "标题名称"
)详细的内容可参考:
22. 相互作用网络图
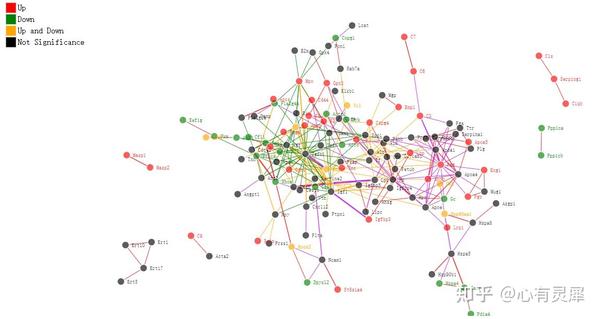
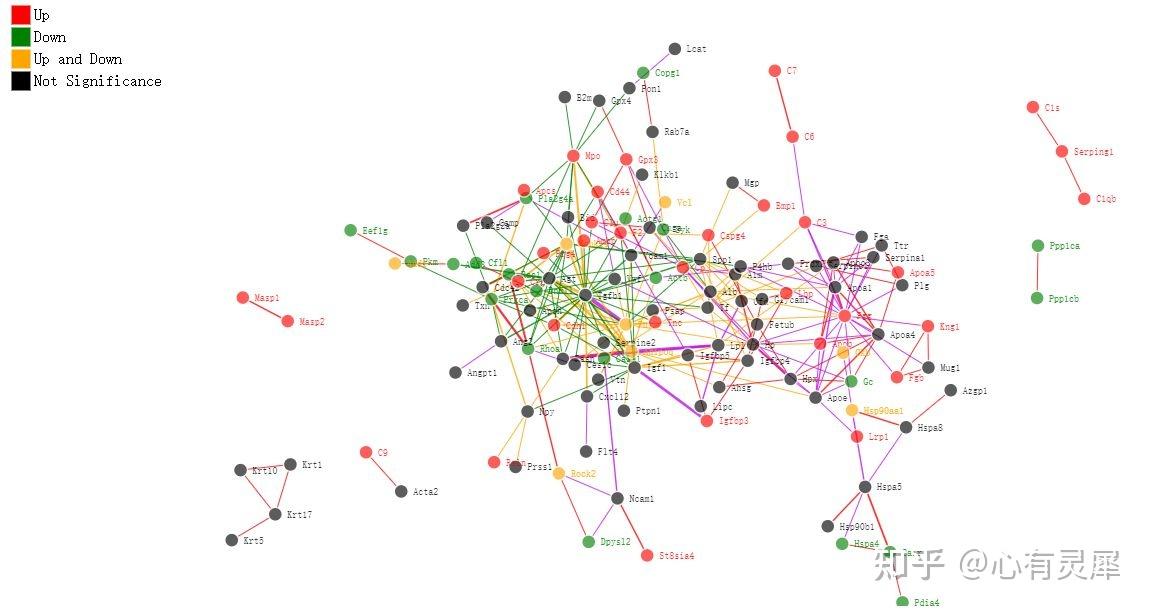
library(tidyverse)
library(networkD3)
# 读取连线文件和节点文件
MisLinks = read.delim("https://www.bioladder.cn/shiny/zyp/bioladder2/demoData/Network/link.csv",sep = ",")
MisNodes = read.delim("https://www.bioladder.cn/shiny/zyp/bioladder2/demoData/Network/node.csv",sep = ",")
# 处理数据
# 因为networkD3需要的连线数据,是节点文件里的名称的索引。所以,需要做一个名称到索引的转化
Node2index = list()
Node2index[MisNodes$name] = 0:length(MisNodes$name)
MisLinks = MisLinks %>%
mutate(source2 = unlist(Node2index[source])) %>%
mutate(target2 = unlist(Node2index[target]))
# 定义颜色
group2project = paste(unique(MisNodes$group),collapse = '","')
color2project = paste(unique(MisNodes$group_color),collapse = '","')
my_color <- paste0('d3.scaleOrdinal().domain(["',group2project,'"]).range(["',color2project,'"])')
forceNetwork(Links = MisLinks,
Nodes = MisNodes,
Source = "source2",
Target = "target2",
Value ="value",
NodeID = "name",
Group = "group",
opacity= 1, # 透明度
Nodesize="size",
zoom = TRUE, # 是否可以缩放
opacityNoHover=1, # 鼠标没有悬浮在节点上时,文字的透明度(0-1)
colourScale = JS(my_color), # 节点颜色,JavaScript
legend=T,
fontSize = 10,
linkColour= MisLinks$colour
)详细的内容可参考:
不想写代码?可以用BioLadder生信云平台在线绘图:
23. 桑基图
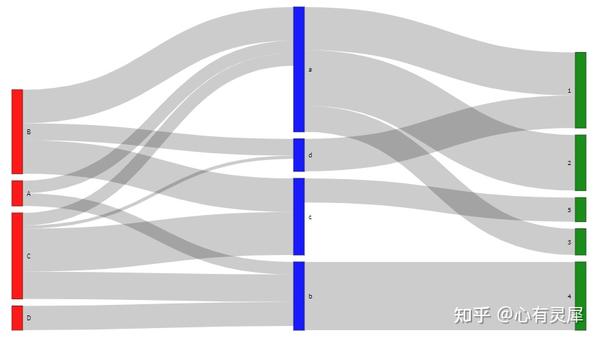
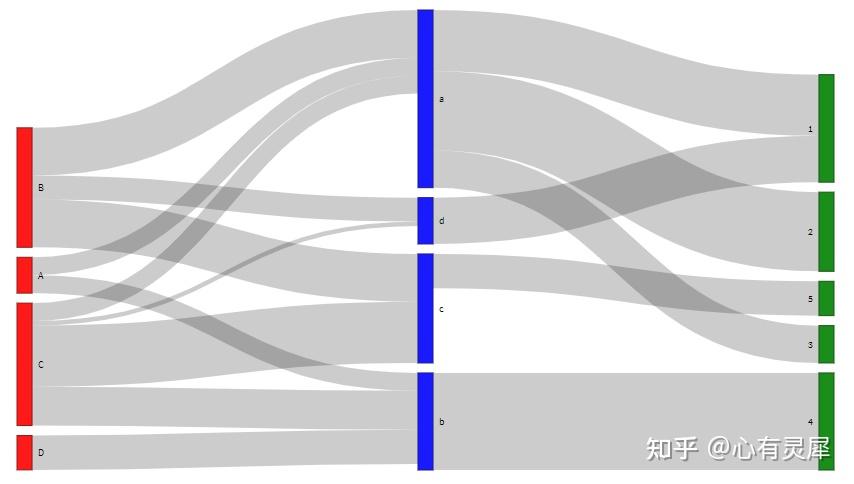
library(networkD3)
library(tidyverse)
# 读取连线文件和节点文件
MisLinks = read.delim("https://www.bioladder.cn/shiny/zyp/bioladder2/demoData/sankeyNetwork/link.csv",sep = ",")
MisNodes = read.delim("https://www.bioladder.cn/shiny/zyp/bioladder2/demoData/sankeyNetwork/node.csv",sep = ",")
# 处理数据
# 因为networkD3需要的连线数据,是节点文件里的名称的索引。所以,需要做一个名称到索引的转化
Node2index = list()
Node2index[MisNodes$name] = 0:length(MisNodes$name)
MisLinks = MisLinks %>%
mutate(source2 = unlist(Node2index[source])) %>%
mutate(target2 = unlist(Node2index[target]))
# 定义颜色
color2project = paste(unique(MisNodes$group_color),collapse = '","')
my_color <- paste0('d3.scaleOrdinal().domain(["',color2project,'"]).range(["',color2project,'"])')
sankeyNetwork(Links = MisLinks,
Nodes = MisNodes,
Source = "source2",
Target = "target2",
Value ="value",
NodeID = "name",
NodeGroup = "group_color",
colourScale = JS(my_color),
fontSize = 10
)详细的内容可参考:
不想写代码?可以用BioLadder生信云平台在线绘图:
24. 散点密度图
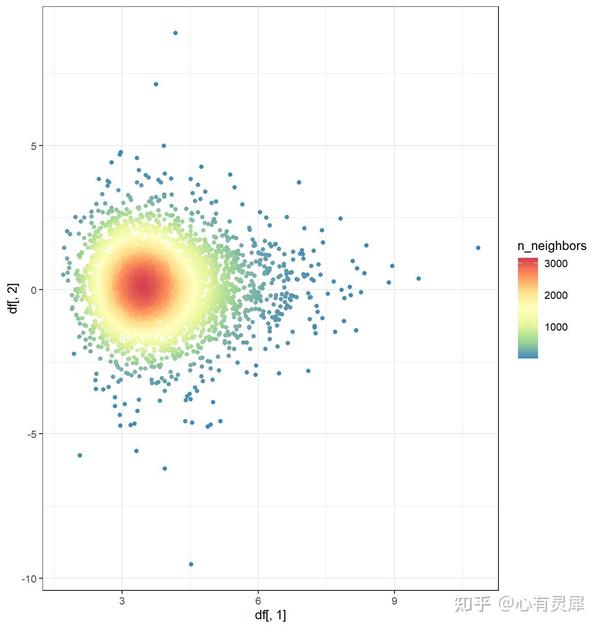
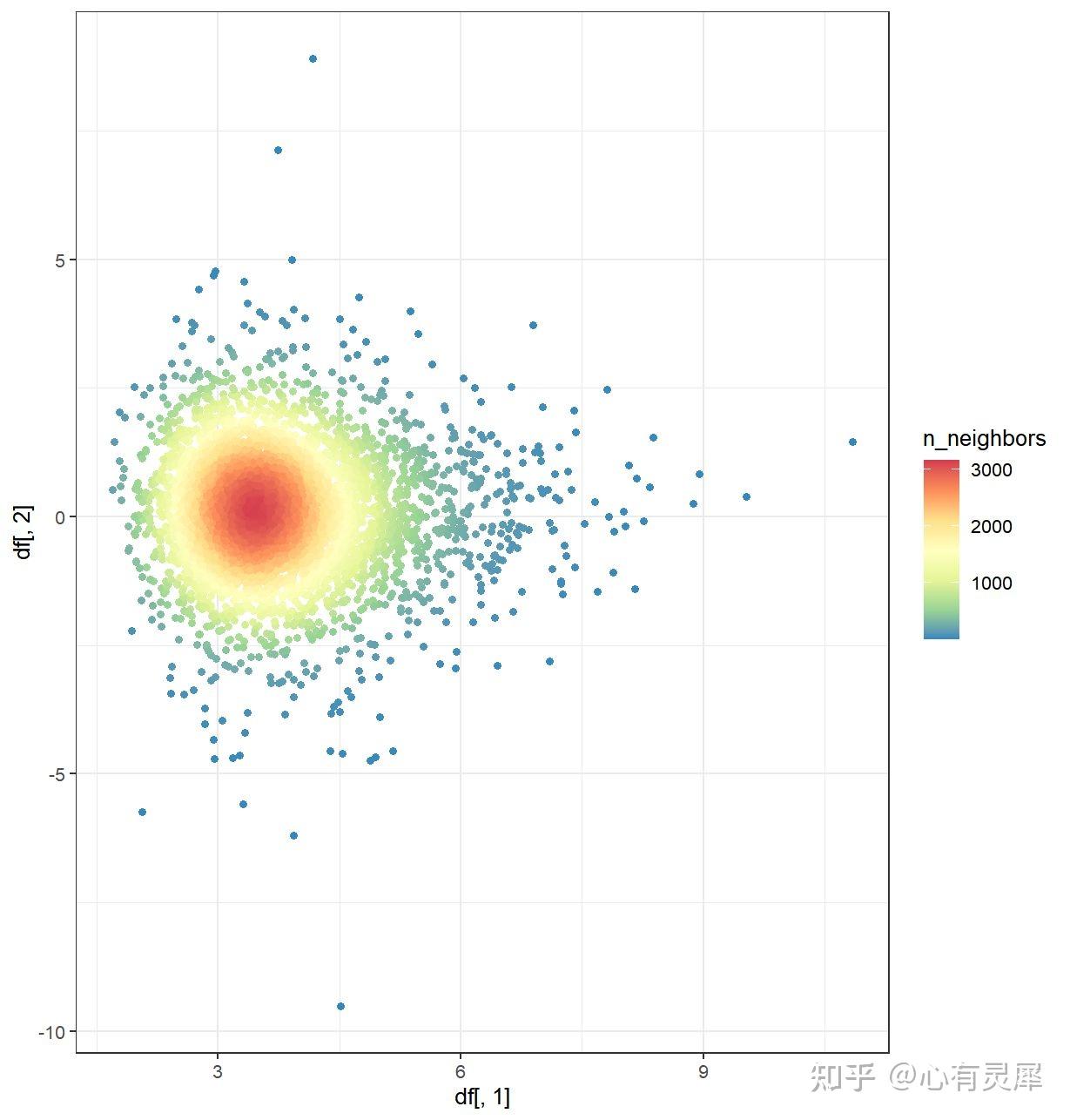
library(ggpointdensity)
library(ggplot2)
# 读文件
df = read.delim("https://www.bioladder.cn/shiny/zyp/bioladder2/demoData/scatterDensity/demo.txt")
ggplot(df,aes(x=df[,1],
y=df[,2])
geom_pointdensity(adjust = 4)+ # adjust:设置neighbors范围
theme_bw()+
scale_color_distiller(palette = "Spectral", direction = -1) # 设置连续型颜色详细的内容可参考:
不想写代码?可以用BioLadder生信云平台在线绘图:
25. PCoA主坐标分析
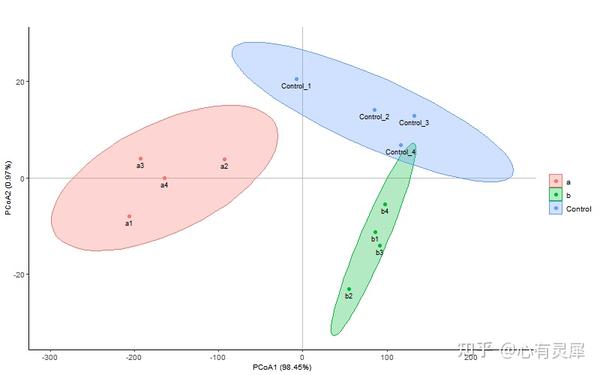
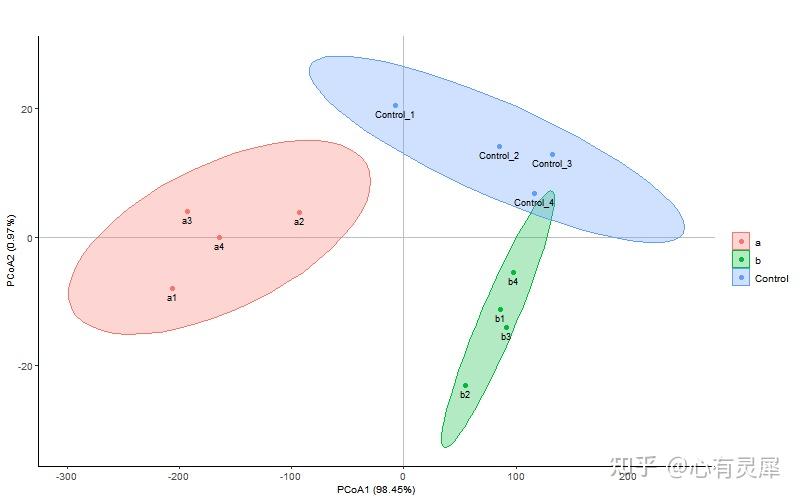
# 加载R包,没有安装请先安装 install.packages("包名")
library(ggplot2)
library(ade4) # 用于计算PcoA
library(vegan) # 用于计算距离
# 读取PCoA数据文件
df = read.delim("https://www.bioladder.cn/shiny/zyp/bioladder2/demoData/PCoA/data.txt",# 这里读取了网络上的demo数据,将此处换成你自己电脑里的文件
header = T, # 指定第一行是列名
row.names = 1 # 指定第一列是行名
df=t(df) # 对数据进行转置,如果想对基因分组则不用转置
# 读取样本分组数据文件
dfGroup = read.delim("https://www.bioladder.cn/shiny/zyp/bioladder2/demoData/PCoA/class.txt",
header = T,
row.names = 1
# PCoA计算
df.dist = vegdist(df,method='euclidean') #基于euclidean距离
pcoa = dudi.pco(df.dist,
scannf = F, # 一种逻辑值,指示是否应该显示特征值条形图
nf=2) # 保留几个维度的坐标信息
# 整理绘图所需的数据
data = pcoa$li
data$name = rownames(data)
data$group = dfGroup$Group
ggplot(data,aes(x = A1,
y = A2,
color = group,
group = group,
fill = group
geom_point()+
theme_classic()+
geom_vline(xintercept = 0, color = 'gray', size = 0.4) + # 在0处添加垂直线条
geom_hline(yintercept = 0, color = 'gray', size = 0.4) +
stat_ellipse(aes(x=A1, # 添加置信区间圈
y=A2,
geom = "polygon",
level = 0.95,
alpha=0.4)+
geom_text( # 添加文本标签
aes(label=name),
vjust=1.5,
size=2,
color = "black"
labs( # 更改x与y轴坐标为pcoa$eig/sum(pcoa$eig)
x = paste0("PCoA1 (",as.character(round(pcoa$eig[1] / sum(pcoa$eig) * 100,2)),"%)"),
y = paste0("PCoA2 (",as.character(round(pcoa$eig[2] / sum(pcoa$eig) * 100,2)),"%)")
)
详细的内容可参考:
不想写代码?可以用BioLadder生信云平台在线绘图:
26. seqlogo序列标识图
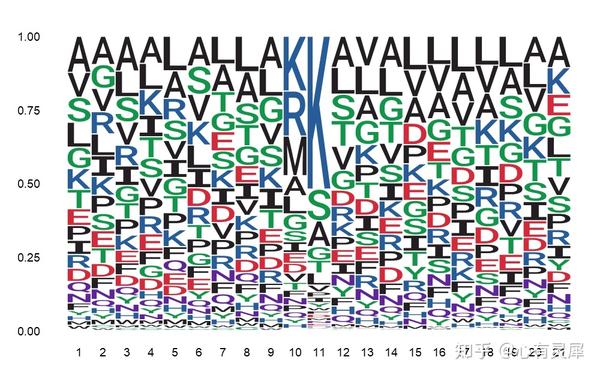
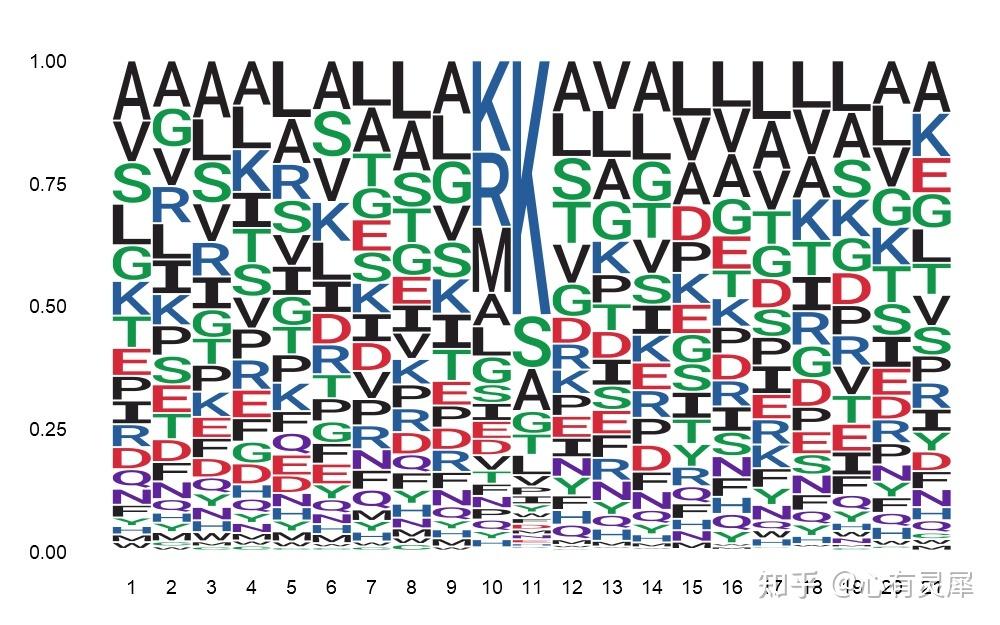
library(ggseqlogo)
df = read.delim("https://www.bioladder.cn/shiny/zyp/bioladder2/model/bioladder1/seqLogo/demoData.txt",header = F)
csl = c()
# # 自定义每个字母的颜色
# csl <- make_col_scheme(chars = c("A"), cols = c("red"))
ggseqlogo(df,
method = "prob", # 定义统计方式"prob"百分比;bits;“custom”
col_scheme = csl) # 定义颜色方案
# 其他基于ggplot2的代码也可以用,略详细的内容可参考:
不想写代码?可以用BioLadder生信云平台在线绘图:
27. 富集弦图
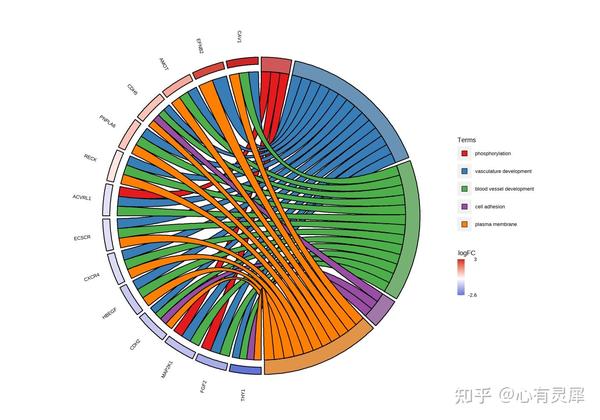
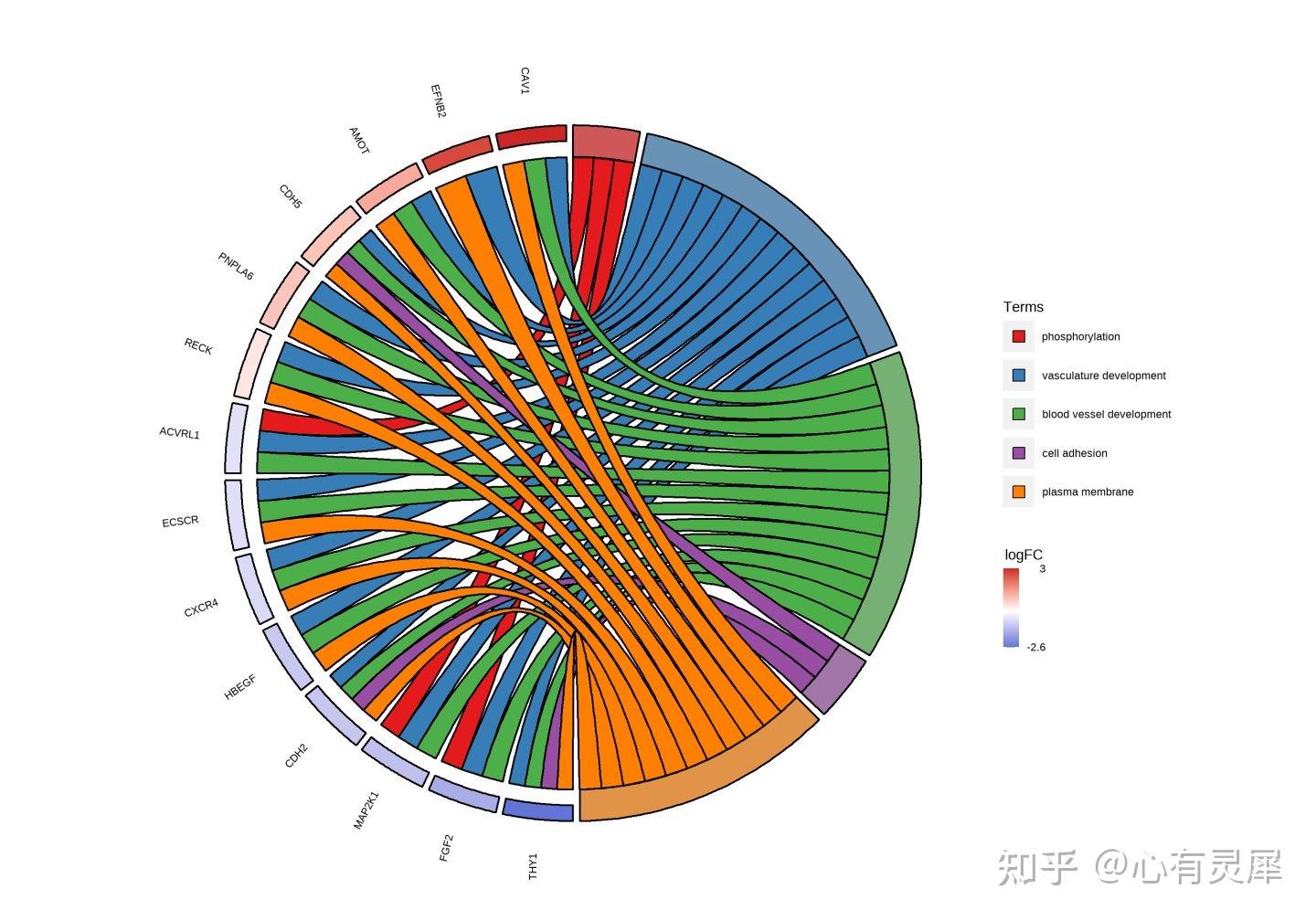
# 转载所需要的包
library(tidyverse) # 用于数据处理
library(GOplot) # 用于绘图 安装方式 devtools::install_github("wencke/wencke.github.io")
library(RColorBrewer) # 颜色
# 读取数据
dfTerm = read.delim("https://www.bioladder.cn/shiny/zyp/bioladder2/model/bioladder1/GOplot/demoData1.txt")
dfFC = read.delim("https://www.bioladder.cn/shiny/zyp/bioladder2/model/bioladder1/GOplot/demoData2.txt")
# 整理数据
dfClean = dfTerm %>%
separate_rows(Genes,sep = ",") %>%
left_join(dfFC,by=c("Genes"="Gene"))
dfPlot = chord_dat(data.frame(dfClean), process=unique(dfClean$Term))
GOChord(dfPlot,
title="GOChord plot", # 标题名称
space = 0.02, # 基因方格之间的空隙
gene.order = "logFC", # 基因的排序方式,可以按照"logFC", "alphabetical", "none",