链接:https://arxiv.org/abs/2006.15222v3
Transformer架构已经被证明可以学习有用的蛋白质分类和生成任务表示。然而,在可解释性方面存在挑战。
本文通过attention分析蛋白质Transformer模型,通过attention(1)捕获蛋白质的折叠结构,将在底层序列中相距很远但在三维结构中空间接近的氨基酸连接起来(2)以蛋白质的关键功能成分结合位点为靶点(3)关注随着层深度的增加而逐渐变得更加复杂的生物物理特性。这一行为在三个Transformer架构(BERT、ALBERT、XLNet)和两个不同的蛋白质数据集上是一致的。并提出了一个三维可视化的方法显示attention和蛋白质结构之间的相互作用。
三.方法论
Model:
Attention analysis:
计算高attention对(
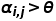 )在数据集X中存在的比例。
)在数据集X中存在的比例。
Datasets:
ProteinNet用于氨基酸和contact map的分析,Secondary Structure用于二级结构的分析,在Secondary Structure的基础上创建了第三个关于结合位点和转录修饰点的数据集,其中添加了从蛋白质数据库web api获得的结合位点和PTM注释。
4.1蛋白质结构
Figure 2
Attention与contact maps在最深层强烈一致:图2显示了根据公式1定义的指标,在被评估的五个模型中,attention如何与contact map相一致。在最深层发现了最一致的头,对接触的关注达到44.7% (TapeBert)、55.7% (ProtAlbert)、58.5% (ProtBert)、63.2% (ProtBert- bfd)和44.5% (ProtXLNet),而数据集中所有氨基酸对的接触背景频率为1.3%。单个头ProtBert- bfd具有最好的效果,其含有420M参数,同时也是唯一在BFD预训练。
考虑到模型是在没有任何空间信息的情况下进行的语言建模任务训练,这些具有结构意识的head的存在值得关注。一种可能是接触更可能发生生物化学作用,在接触的氨基酸之间产生统计依赖。
4.2结合位点和转录修饰点
Figure 3
在模型的大多数层Attention意在结合位点:对结合位点的关注在ProtAlbert模型中最为显著(图3b),该模型有22个头,将超过50%的注意力集中在结合位点上,而数据集中结合位点的背景频率为4.8%。三种BERT模型(图3a、3c和3d)对结合位点的关注也很强,注意头对结合位点的关注分别达到48.2%、50.7%和45.6%。
ProtXLNet(图3 e)目标结合位点,但不像其它模型强烈:最一致的头有15.1%attention关注结合位点,平均头将只有6.2%的attention关注结合位点,而前四个模型均值以次为13.2%,19.8%,16.0%,和15.1%。目前还不清楚这种差异是由于架构的差异还是由于预训练目标的差异;例如,ProtXLNet使用双向自回归预训练方法(见附录a .2),而其他4个模型都使用掩码语言建模。结合位点是蛋白质与其他大分子的相互作用位置,这决定了蛋白质的高级功能,即使序列整体进化,结合位点也将被保留,同时结合位点的结构也局限于特定的家族或超家族,结合位点可以揭示蛋白质之间的进化关系,因此结合位点可能为模型提供对个体序列变化具有鲁棒性的蛋白质的高级描述。
一小部分Head的attention意在PTMs, TapeBert中的Head 11-6集中了64%的注意力在PTM位置上,尽管这些只发生在数据集中0.8%的序列位置上。
4.3跨层分析
在较深层attention意在高级属性:图4中较深的层相对更关注结合位点和contact(高级概念),而二级结构(低级到中级概念)则更均匀地跨层定位;Attention probe显示关于contact map的知识主要在最后1-2层被编码进注意权重,这与基于文本的Transformer模型在较深层次处理更复杂的属性相一致;Embedding probe(图5,橙色)也表明,模型首先在较低层构建局部二级结构的表示,然后在较深层完全编码结合位点和接触图。然而,这一分析也揭示了在接触图的知识如何在嵌入中积累的明显差异,embedding是在多个层次上逐渐积累这种知识,而attention权重则只在最后的层次上获得这种知识。
4.4氨基酸和可替代矩阵
根据图6,attention head关注特定氨基酸,那么每个head是否记住了特定的氨基酸或者学会了与氨基酸相关的有意义的特性,为了验证这个猜想,计算了所有不同氨基酸对与头部注意力分布之间的皮尔逊相关系数(图7 左)并发现与BLOSUM62(图7 右)的皮尔逊系数为0.73,表明attention适度与可替代关系统一。
本文将NLP的可解释性方法应用于蛋白质序列建模,并在此基础上建立了NLP与计算生物学的协同效应并展示了Transformer语言模型如何恢复蛋白质的结构和功能特性,并将这些知识直接整合到它的注意机制中。虽然本文的重点是将注意力与已知的蛋白质特性协调起来,但人们也可以利用注意力来发现新的关系或现有措施的更细微的形式
OpenKG
开放知识图谱(简称 OpenKG)旨在促进中文知识图谱数据的开放与互联,促进知识图谱和语义技术的普及和广泛应用。
点击
阅读原文
,进入 OpenKG 网站。
原文链接:
http://openkg.cn
此项目尚无适当的setup.py/pip安装,因此只需克隆该项目,然后将protein
bert
子目录添加到您的
python
路径
中
即可(例如,通过修改
PYTHON
PATH virtualenv或使用sys.path.append)。
下载,了解如何在新的
蛋白质
数据集上微调预训练的Protein
bert
模型
。
更新Protein
BERT
demo.ipynb
使基准文件可通过FTP访问
创建setup.py和pip安装
编写适当的文档(包括安装说明和依赖项,用法,有关该软件包的一些背景以及如何引用它)。
请注意,依赖项包括tensorflow_addons
包括有关如何从头开始创建数据集以及如何对
模型
进行预训练的说明(请参见下文); 如有必要,将数据集文件添加到FTP。
如果保持原样,请更新original_dataset.h5
中
的“ included_annotat
Atomic protein structure refinement using all-atom graph representations and SE(3)-equivariant graph transformer
来源:知乎(ID:熊墨淼)最近两星期(7月15日,7月22日),《自然》杂志连续发表两篇DeepMind写的用
人工智能
的方法预测
蛋白质
三维结构的文章和NIH Director Franci...
根据使用方法,将按照以上顺序对每部分代码进行分析。
特征提取 : Embedding Section
ProtTrans/Embedding/Onnx/Prot
Bert
-BFD.ipynb
ONNX简介:
Open Neural Network Exchange(ONNX,开放神经网络交换)格式,是一个用于表示深度学习
模型
的标准,可使
模型
在不同框架之间进行转移。ONNX是一种针对
机器学习
所设计的开放式的文件格
如果想进一步深入研究,则需要了解Transformers库
中
更底层的实现,学会对具体的
BERT
o
log
y系列
模型
进行单独加载和使用。
1 Transformers库的文件结构
1.1 详解Transformers库
中
的预训练
模型
在Transformers库
中
,预训练
模型
文件主要有3种,它们的具体作用如下:
词表文件:在训练
模型
时,将该文件当作一个映射表,把输入的单词转换成具体数字。(文本方式保存)
配置文件:存放
模型
的超参数,将源码
中
的
模型
类根据配置文件的超参数进行实例化后生成可用的
模型
。(..
目录1. 研究背景2. 研究数据2.1 预训练的
蛋白质
数据集2.2
蛋白质
基准数据集3. 研究方法3.1 序列和标注编码3.2
蛋白质
序列和注释的自我监督预训练3.3 对
蛋白质
基准进行监督微调3.4 深度学习框架4. 结果4.1 预训练可以改善
蛋白质
模型
4.2 Protein
BERT
在不同的
蛋白质
基准上达到了近乎最先进的结果4.4 全局
注意力
机制的理解5. 结论
作者单位:耶路撒冷希伯来大学
发表期刊:《Bioinformatics》,2020年期刊影响因子:6.937
发表时间:2022年1月9日
细调: Fine Tuning Section
import torch
from transformers import AutoTokenizer, Trainer, TrainingArguments, AutoModelForTokenClassification,
Bert
TokenizerFast, EvalPrediction
from torch.utils.data import Datase
人类基因组编码超过500种不同的蛋白激酶,它们通过
蛋白质
底物的特定磷酸化来调节几乎所有的细胞过程。虽然质谱和
蛋白质
组学研究的进展已经确定了跨物种的数千个磷酸化
位点
,但对于绝大多数磷酸化
位点
,目前缺乏关于磷酸化这些
位点
的特定激酶的信息。最近,预测激酶-底物关联的计算
模型
的发展得到了广泛的关注。然而,目前的
模型
只允许对研究充分的激酶子集进行预测。此外,在训练和测试数据集
中
利用手工处理的特征和不平衡,对开发激酶特异性磷酸化预测的准确预测
模型
提出了独特的挑战。
文章目录一、摘要二、方法(一)、
蛋白质
图的表示(二)、节点级特征表示(三)、边级特征表示(四)、 EGAT结构的主要特点(五)、边缘聚合图关注层(六)、在计算
注意力
分数时使用边缘特征三、结果(一)、数据集(二)、基准数据集上的结果(三)使用基于 Prot
BERT
的特性进行迁移学习的影响四、总结
图神经网络(GNN)已成为结构信息编码的一种有效工具,尽管基于GNN的体系结构已被应用于配对
结合
位点
预测,但他没有被用来预测单个
蛋白质
的
结合
位点
。此外,与可能不适当编码远程相互作用的残基特异性信息的方法不同
通过使用KGs来辅助数据驱动的药物研究,可以加速药物发现的过程。受此应用的启发,最近的研究将KG引入到基于KG嵌入
模型
和生物医学KG的各种药物发现预测问题
中
。在本文
中
,我们总结了用于KG构建的常用数据库,并概述了药物发现领域
中
具有代表性的知识嵌入
模型
和基于KG的预测。最后,我们谨慎地总结,未来仍有一些挑战需要解决,并对这些挑战进行了详细的讨论,旨在为该领域的未来提供清晰的前景。
Prediction Section
ProtTrans/Prediction/Prot
Bert
-BFD-Predict-SS3.ipynb
from transformers import AutoTokenizer, AutoModelForTokenClassification, TokenClassificationPipeline
import re
pipeline = TokenClassif
SARS-COV-2号召科学界采取行动,以对抗日益增长的大流行病。撰写本文时,还没有新型抗病毒药或批准的疫苗可用于部署作为一线防御。了解COVID-19的病理
生物学
特性可通过阐明未探索的病毒途径来帮助科学家发现有效的抗病毒药。实现这一目标的一种方法是利用计算方法在计算机上发现新的候选药物和疫苗。过去的十年
中
,基于
机器学习
的
模型
在特定的生物分子上进行了训练,为发现有效的病毒疗法提供了廉价且快速的实施方法。给定目标生物分子,这些
模型
能够以基于结构的方式预测候选抑制剂。如果有足够的数据提供给
模型
,则可以通过识别.

