

如何打开gtf格式的gencode基因注释文件(记事本 vs R语言rtracklayer)

gencode网站提供了基因注释文件
1 打开方式:记事本-excel
小一点的gtf格式文件可以当成txt格式用文记事本打开,然后复制粘贴到excel
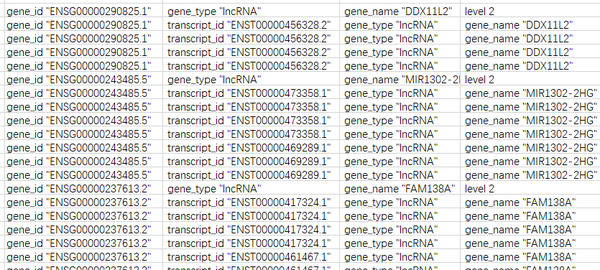
如lncRNA注释文件:
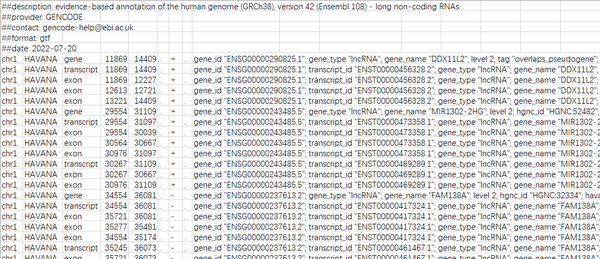
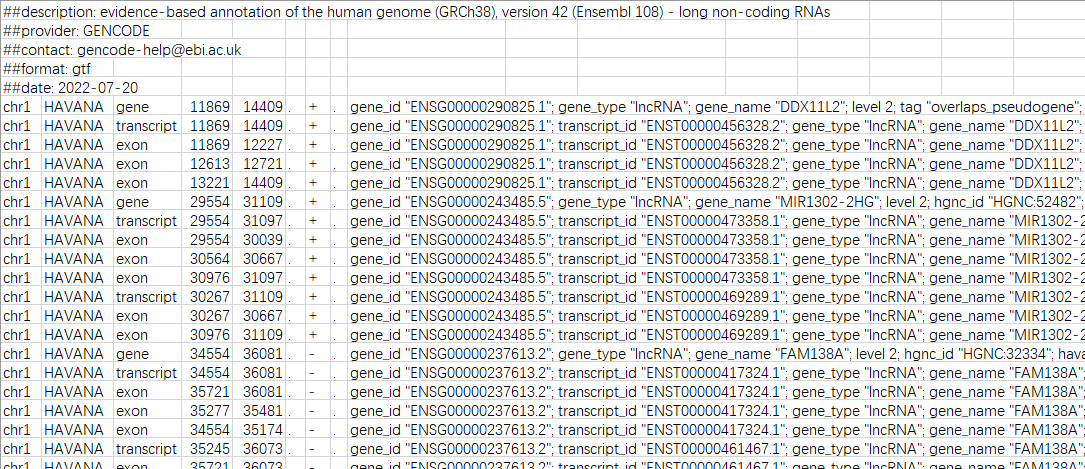
虽然格式不太完美(后面的gene_id、gene_name等都没分开),在excel调一下也还是可以的:
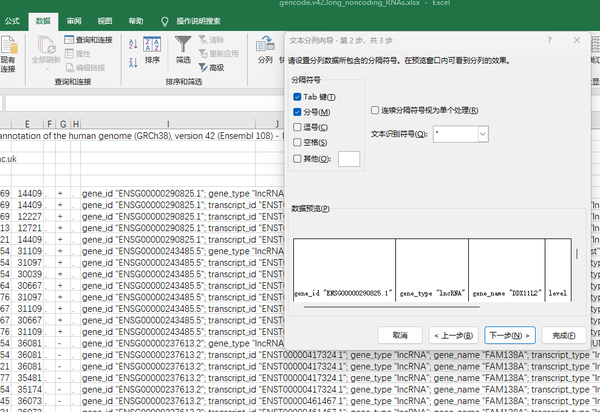
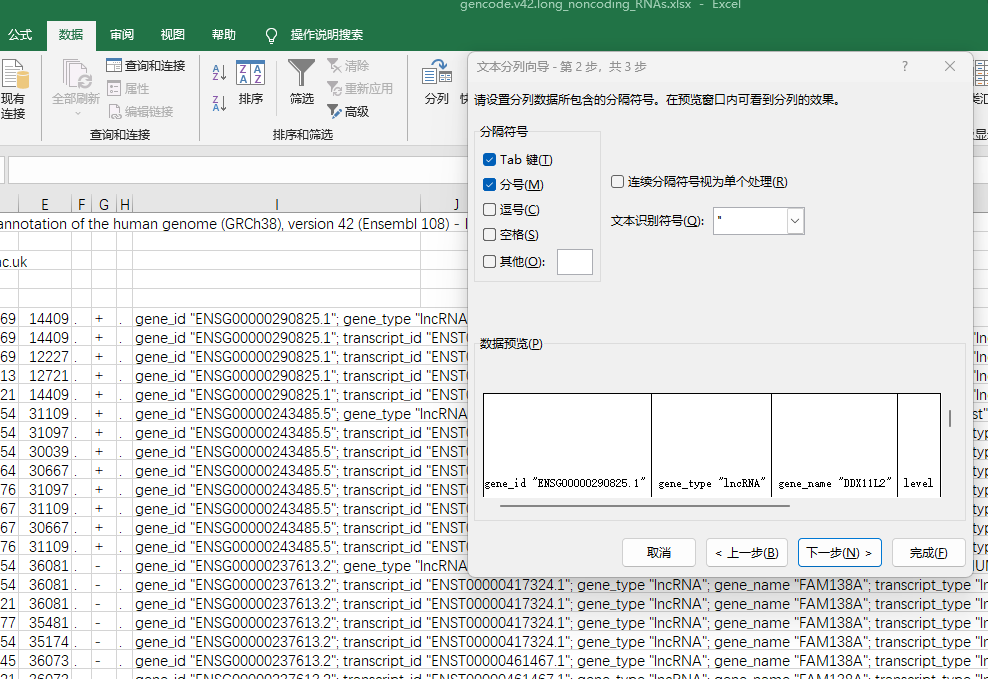
选定数据分的不好的这一列,在“数据”栏点击“分列”,选择“分隔符号”分列,然后选上“分号”,这时候分号间隔的就分开了。
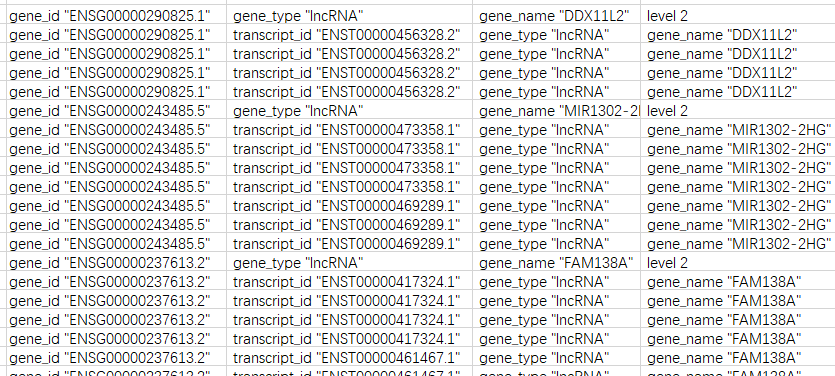
里面多的字符如gene_id手动删除
2 打开方式:R语言rtracklayer
太大的注释文件用记事本就打不开了,可以用R语言中的rtracklayer包打开:
library("rtracklayer") #加载rtracklayer包
gc_data = import('gencode.v42.annotation.gtf') #输入要打开的gtf注释文件
gc_data <- as.data.frame(gc_data)#将文件转换为数据框格式这时候就得到了数据:
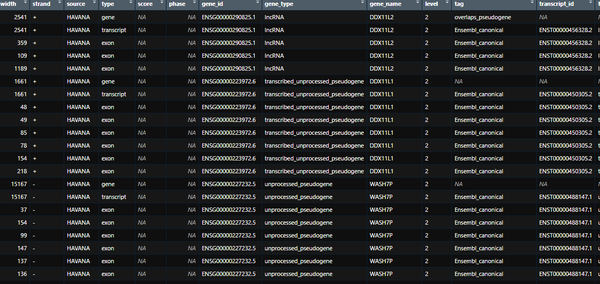

数据已完全分开,还没有多的字符。
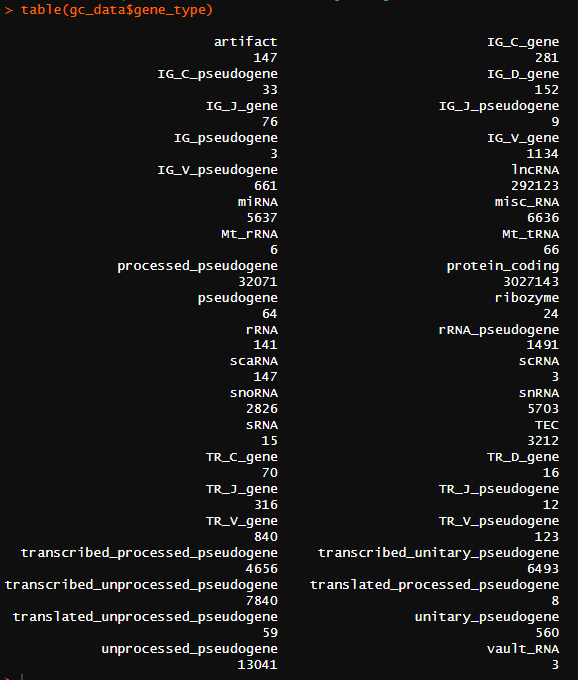
看一下有多少gene类型:
table(gc_data$gene_type)
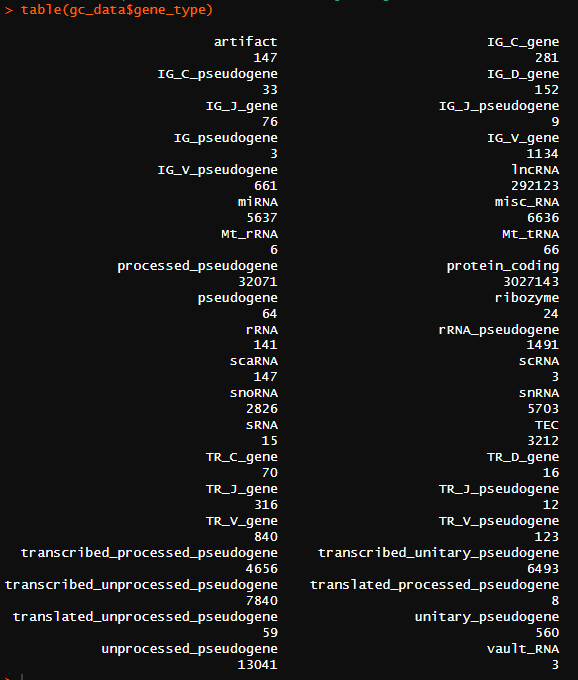
3 输出
可以直接输出为csv表格:
write.csv(gc_data,file = "gc.csv")太大,不建议直接输出。
可以筛选出自己需要的数据再输出
如筛选出基因型为lncRNA和pseudogene的基因
3.1 先把基因名带“pseudogene”的基因名提取出来
lncRNA只有一种,但是pseudogene有很多种,所以需要先把基因名中带“pseudogene”的基因名提取出来
gt <- as.data.frame(table(gc_data$gene_type)) # 转换成数据框
gtp <- gt$Var1[str_detect(gt$Var1,"pseudogene")] # 提取出名称中带pseudogene的基因名称