


DeepProg:使用多组学数据进行预后预测的深度学习和机器学习模型的集合

Genome Medicine (2021)
Olivier B. Poirion
,
Zheng Jing
,
Kumardeep Chaudhary
,
Sijia Huang
&
-
Lana X. Garmire
密歇根大学计算医学和生物信息学系
多组学数据是预后和生存预测的良好资源;然而,这些很难在计算上集成。我们介绍了 DeepProg,这是一种新的深度学习和机器学习方法的集成框架,它使用多组学数据稳健地预测患者生存亚型。它确定了大多数癌症中的两种最佳生存亚型,并且比其他多组学整合方法产生了明显更好的风险分层。DeepProg 具有高度预测性,例如两个肝癌(C-index 0.73-0.80)和五个乳腺癌数据集(C-index 0.68-0.73)。泛癌分析将不良生存亚型中的常见基因组特征与细胞外基质建模、免疫失调和有丝分裂过程联系起来。DeepProg 可 免费获得 在
大多数基于生存的分子特征都基于一种单一类型的组学数据 [ 1 ]。由于每个组学平台都有特定的限制和噪声,基于多组学的综合方法可能会产生更连贯的特征[ 2 ]。然而,由于计算和实际挑战的结合,这种预测临床表型的方法相对较少被探索。这些挑战包括特定于平台的测量偏差 [ 3 ]、需要适当归一化的不同数据分布 [ 4 ],以及由于成本高而导致的多组学测量的样本量非常有限 [ 5 ]。
在多组学数据集成方法中,大多数都没有以患者生存为目标;相反,与分子亚型相关的生存差异是以事后的方式评估的 [ 6 ]。此外,许多方法使用无监督方法,不适合预测新的患者状态 iCluster 、相似性网络融合 (SNF)、MAUI 和多组学因子分析 (MOFA+) 就是这样的例子 [ 7 , 8 , 9 , 10 ]。iCluster 是最早基于多组学特征将癌症样本聚类为不同分子亚型的方法,使用概率建模将数据投影到较低的嵌入 [ 7 ]。相似性网络融合 ( SNF ) 算法是另一种流行的聚类方法,用于集成不同的组学特征,首先为每个组学构建一个不同的相似性网络,然后使用迭代过程融合网络 [ 8 ]。它被应用于多个 TCGA 癌症数据集 [ 11 , 12 ]。 MAUI 是一种用于多组学集成的非线性降维框架,它使用变分自动编码器来生成可用于聚类或分类的潜在特征 [ 9 ]。同样, MOFA+ 是一个统计框架,它通过标准矩阵分解使用因子分析来从多组学数据集中推断潜在变量,从而解释最大的变异来源 [ 10 ]。
识别疾病亚型在临床上非常重要 。例如,普遍认为特定器官的癌症具有多种亚型。使用分子特征来识别癌症亚型允许肿瘤分类超出肿瘤阶段、等级或起源组织 [ 13 ]。 具有相似分子和通路改变的癌症亚型可以用相同的药物治疗 [ 14 ]。其中一种亚型是基于 预后特征的生存分层患者亚型 [ 15 ]。一旦推断出来,它们的特征就可以作为后续治疗或预后研究的起点 [ 16 ]。此外,与患者生存相关的分子差异有助于了解 肿瘤进展的机制 [ 17 ]。获得的知识不仅有助于 改善疾病监测和管理 ,而且为 预防和治疗 提供信息。
在这里,我们提出了一个独特的计算建模框架,称为 DeepProg,不同于上述所有方法。它明
确地将患者生存建模为目标,并预测新患者的生存风险
。DeepProg 构建了一个灵活的混合模型集成(深度学习和机器学习模型的组合),并按照集成学习范式集成它们的输出。我们将 DeepProg 应用于癌症基因组图谱 (TCGA) 中 32 种癌症的
RNA-Seq、甲基化和 miRNA 数据
,总共大约 10,000 个样本。与基于 SNF 的多组学集成方法和基线 Cox-PH 方法相比,DeepProg 显示出更好的预测准确性。所有癌症中
生存最差的亚型中的基因表达具有共同的特征
,这些特征涉及生物学功能,例如
有丝分裂增强,细胞外基质不稳定或免疫失调
。此外,DeepProg 可以使用基于其他癌症的模型成功预测来自一种癌症的样本的结果。简而言之,DeepProg 是一种强大的、通用的、基于机器学习和深度学习的方法,可用于预测个体患者的生存亚型。
DeepProg 方法概述
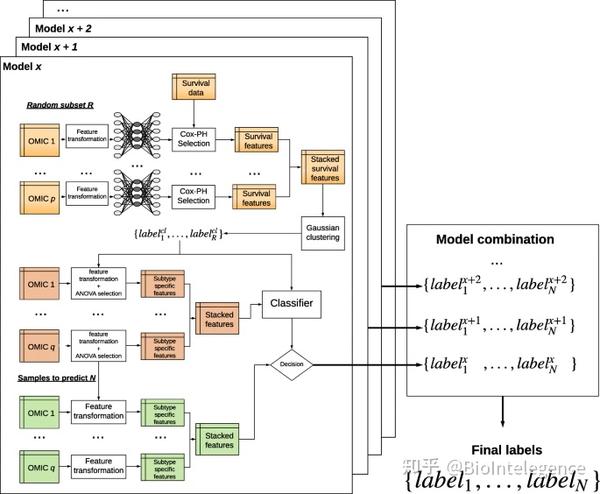
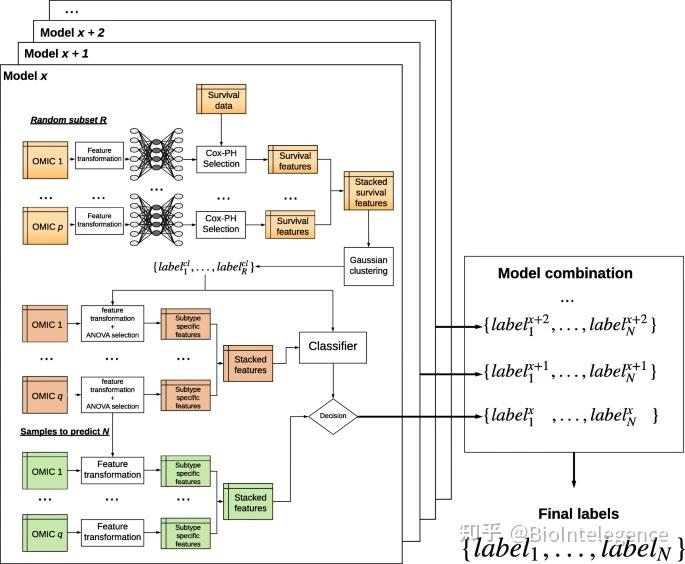
DeepProg 是一个通用的混合和灵活的计算框架,用于根据一种或多种组学数据类型预测患者的存活率,例如 mRNA 转录组学、DNA 甲基化和 microRNA 表达(图 1 ) )。DeepProg 的第一阶段由使用自定义秩归一化和自动编码器(一种深度神经网络)的降维和特征转换组成。它对自动编码器使用“模块化”设计,其中每种数据类型都由一个自动编码器建模,以实现异构数据类型的灵活性和可扩展性。在默认实现中,自动编码器有 3 层,输入层、隐藏层(100 个节点)和输出层。然后对转换后的特征进行单变量 Cox-PH 拟合,以进一步选择与生存相关的特征子集。接下来,使用无监督聚类方法,DeepProg 识别生存亚群的最佳类别(标签)数量,并使用这些类别构建基于支持向量机(SVM)的机器学习模型,以预测新患者的生存组。为了确保模型的鲁棒性,DeepProg 采用了 boosting 方法并构建了一个模型集合。boosting 方法产生更准确的 p 值和 C 指数,方差更小,并导致模型更快收敛(附加文件 1 :表 S1)。这些模型中的每一个都是用原始数据集的随机子集(例如,4/5)构建的,并使用来自剩余保留(例如,1/5)测试样本的 C 指数值进行评估。为了提高效率,DeepProg 的计算是完全分布式的,因为每个模型都可以单独拟合。
32 种 TCGA 癌症的预后预测
我们应用 DeepProg 分析了 TCGA 中 32 种癌症的多组学数据(RNA-Seq、miRNA-Seq 和 DNA 甲基化)(附加文件 2 :表 S2)。我们仅对卵巢癌 (OV) 使用 RNA 和 MIR,因为在提交手稿时只有一小部分样本(300 个样本中的 9 个)具有 3 组学数据。对于每种癌症类型,我们选择了最佳聚类数 K,它产生轮廓分数和调整后的兰德指数(附加文件 3 :表 S3)的最佳组合,衡量聚类稳定性和准确性的指标。几乎所有癌症(32 种癌症中的 30 种)都将 K = 2 作为最佳生存亚组(图 2 一个)。使用最佳聚类数,我们计算了每种癌症不同生存亚型之间的对数秩 p 值,所有这些都具有统计学意义(对数秩 p 值 < 0.05)并且 C 指数(0.6-1.0)大于0.5,随机模型的期望值。其中,32 种癌症中有 23 种的对数秩 p 值小于 5e-4,突出了模型在区分患者生存率方面的价值(图 2B )。此外,我们针对每种组学数据类型和每种癌症调查了与生存显着相关的隐藏层特征的平均数量(附加文件 4 :图S1)。总体而言,RNA-Seq 在生存预测方面具有最多的重要隐藏特征。miRNA 隐藏特征在所有癌症中具有相似的模式,总计数较少。尽管 32 种癌症之间存在巨大的异质性,但一些已知密切相关的癌症,如结肠癌 (COAD) 和胃癌 (STAD),以及膀胱癌 (BLCA) 和肾癌 (KIRC),也具有相似的预后隐藏特征。
我们之前表明,对于 DeepProg 的原型,添加癌症分期、种族等临床变量无助于提高 HCC 的预测结果 [ 18 ]。在这里,我们还直接比较了 DeepProg 与基于阶段分层(I + II 期与 III + IV 期)的简单模型对 32 种癌症类型的性能。如附加文件 4所示 :图 S2A,与基于阶段分层生存差异的简单模型相比,DeepProg 具有显着更好的(秩和检验 p 值 = 2.4e−3)对数秩 p 值。为了进一步证明 DeepProg 能够预测超过肿瘤阶段的患者生存结果,我们接下来关注晚期(III 和 IV 期)COAD 和 STAD。我们使用后期 III 和 IV 的患者构建了 DeepProg 模型,并使用 III 期和 IV 期将其结果与生存差异进行了比较(附加文件 4 : 图 S2B-E)。DeepProg 确定的 STAD 和 COAD 亚型明显比基于肿瘤分期的亚型更显着(对数秩 p 值分别为 5.5e-04 和 2.7e-06)(对数秩 p 值分别为 0.16 和 0.012)。此外,来自 DeepProg 的亚型与分期没有显着相关性(COAD 和 STAD 的 Fisher 精确 p 值分别为 0.08 和 0.14)。因此,DeepProg 提供了比临床因素肿瘤分期更多的信息来预测患者的存活率。
DeepProg 与其他方法的比较
为了评估新的 DeepProg 方法,我们将上述 32 种癌症的结果与从相似性网络融合 (SNF) 算法 [ 8 ] 中获得的结果进行了比较,这是一种整合多组学数据的最先进方法(图 1)。 2 B,C,附加文件 4 :图 S3)。以前 SNF 被用于识别与其他人的生存相关的癌症亚型 [ 11 , 12 ]。如图 2B 所示,来自 SNF 的生存 亚型 仅在 32 种癌症中的 13 种具有显着的生存差异(p 值<0.05)。总之,DeepProg 产生了更好的对数秩 p 值(图 2 B)和 C 指数(图 2 C)。此外,考虑到自 SNF 发布以来 TCGA 数据集可能已经发生变化,我们还在五个测试数据集上使用了原始 SNF 论文中确定的患者亚型,并使用它们来获得对数秩生存亚型 p 值 [ 8 ]。与使用相同的五个数据集作为输入从 DeepProg 获得的值相比,这些 p 值都不太显着(附加文件 5 :表 S4)。
我们还使用 TCGA HCC 和 BRCA 数据集,用简单的 PCA 分解和两种矩阵分解方法(包括 MAUI 和 MOFA+)替换了 DeepProg 配置的自动编码器步骤。在每种替代方法中,我们将多组学矩阵转换为 100 个新组件,然后在 DeepProg 中执行相同的剩余步骤(例如,与生存相关的特征过滤、聚类)。虽然 MAUI 的 100 个特征在 Cox-PH 过滤步骤中的 ap 值均不小于 0.05,但与 DeepProg 中的默认降维步骤相比,PCA 和 MOFA+ 的性能都差得多(图 3 和附加文件 6 :表 S5)。在 HCC 检测数据上,PCA 和 MOFA+ 的 C 指数分别为 0.60 和 0.59(图 3 A,B),而 DeepProg 的结果为 0.76(图 3 C)。在 BRCA 测试数据中,PCA 和 MOFA+ 的 C 指数分别为 0.58 和 0.62(图 3 D、E),而 DeepProg 的 C 指数为 0.77(图 3 F)。总之,基于 HCC 和 BRCA 基准,与矩阵分解方法和 PCA 相比,DeepProg 从多组学数据推断与生存相关的亚型明显更好。
最后,我们将 DeepProg 与基线模型进行了比较,其中 Z-score 归一化特征由 Cox-PH 模型直接拟合,带有 Lasso 惩罚,无需自动编码器步骤。样本被分成与 DeepProg 中相同数量的集群,只要适用,就对相同的参数化进行主观判断。在 32 种癌症的相同保留样本上,DeepProg 显示出比基线 Cox-PH 模型显着更好的对数秩 p 值(p 值 < 0.0005,2 边 t 检验)(附加文件 4 :图 S4)。
其他群组对 DeepProg 性能的验证
DeepProg 工作流程的一个关键优势是它能够预测任何新的个体样本的生存亚型,这些样本具有一些与训练数据集相同的 RNA、miRNA 或 DNA 甲基化特征(图 1B )。DeepProg 通过获取特征的相对等级来归一化新样本,并使用它们来计算到训练集中样本的距离(参见“ 方法 ”部分)。为了验证 DeepProg 模型的患者生存风险分层,我们将它们应用于其他独立的癌症数据集,两个来自肝细胞癌 (HCC) 队列(图 4 A、B)和四个来自乳腺癌 (BRCA) 队列(图 4 C、F)。两个 HCC 验证集是具有 230 个 RNA-Seq 样本的 LIRI 数据集和具有 221 个基因表达阵列结果的 GSE 数据集(参见“ 方法 ”部分)。我们分别获得了 0.80 的 C 指数和 1.2e-4 (LIRI) 的对数秩 p 值和 0.73 的 C 指数和 1.5e-5 (GSE) 的对数秩 p 值(图 4 A ,乙)。四个 BRCA 数据集的 C 指数为 0.68-0.73,对于生存差异都有显着的对数秩 p 值(<0.05)(图 4 C、F)。因此,我们通过额外的 HCC 和 BRCA 队列验证了 DeepProg 的可预测性。
鉴定最差生存亚型的特征基因揭示了泛癌模式
为了确定与患者生存差异相关的关键特征,我们对每个组学层中的特征进行了综合分析,这些特征在生存最差的患者子集中显着过度或过度表达。接下来,在表达过度或表达不足的特征中,我们从 Wilcoxon 秩检验 p 值小于 1e-4 的输入数据类型中选择重要特征。对于这些特征中的每一个,我们计算了每种癌症类型的单变量 Cox-PH 回归,并根据 -log10(p 值)对它们进行排序。在将这些等级在 0 和 1 之间标准化后,我们通过对所有 32 种癌症类型求和来获得泛癌等级(参见“ 方法 “ 部分)。 我们在下面描述了 RNA-Seq 分析的结果,并在附加文件7 中总结了 microRNA 和 DNA 甲基化分析的结果。
RNA-Seq 分析显示了在最差的生存组中出现了一些过度表达的基因模式(图 5A )。CDC20 排名第一,其他一些来自细胞分裂周期 (CDC) 家族的基因,包括 CDCA8、CDCA5、CDC25C 和 CDCA2,也在前 100 位基因之列(附加文件 8 :表 S6)。此外,前 100 个基因中存在大量来自驱动蛋白家族成员 (KIF) 的基因(即 KIF4A、KIF2C、KIF23、KIF20A、KIF18A、KIFC1、KIF18B 和 KIF14)(附加文件 8 :表 S6)。CDC 基因 [ 31 , 32 , 33 , 34 , 35 ] 和 KIF 基因过度表达 [ 36 , 37 ] 已在转移过程中被报道并与预后不良有关。许多其他在低生存率组中过度表达的基因与之前的研究一致,例如 ITGA5、CALU、PLKA1、KPNA2、APCDDL1、LGALS1、GLT25D1、CKAP4、IGF2BP3 和 ANXA5 [ 38 ]。使用排名值,我们对癌症和基因进行聚类,并检测到两个清晰的基因簇,富含细胞周期和有丝分裂的生物学功能(Adj. p 值 = 3e-42)和细胞外基质组织途径(Adj. p 值 = 6e-9),分别(图 5A )。此外,分析显示了两组不同的癌症,其中 GBM、HNSC、OV、STAD、COAD、LUSC 和 KIRC 属于一组,而 PRAD、PAAD 和 LUAD 等癌症属于另一组(图 1)。 5 A)。
在最差生存组中表达不足的基因中,CBX7 和 EZH1 是前 2 个基因(附加文件 8 :表 S6)。已证明 CBX7 的下调在癌症进展中起关键作用 [ 39 ]。同样,EZH1 抑制被证明与细胞增殖和致癌作用有关 [ 40 , 41 ]。此外,锌指家族中的多个基因被下调(ZBTB7C、ZMAT1、ZNF18、ZNF540、ZNF589、ZNF554 和 ZNF763)。ZNF 基因是转录因子的一个大家族,其中许多与癌症进展有关[ 42 ]。
RNA-Seq 共表达网络分析
为了进一步表征与最差生存亚型相关的 RNA-Seq 基因表达,我们进行了 全局基因共表达分析 。对于每种癌症类型,我们从最差生存亚型中选择了差异表达的基因(图 5A ),并构建了一个泛癌共识共表达网络。作为说明,我们使用前 200 个基因和最重要的边缘(图 5 B)构建了一个共表达子图,并使用随机游走算法进行了基因群落检测 [ 43 ]。大部分顶级共表达基因与前面强调的顶级生存基因重叠。例如,一个紧密簇(第 1 组)由多个 CDC 和 KIF 基因以及 BUB1、MCM10、AURKB、CENPA、CENPF 和 PLK1 组成。这些基因与 有丝分裂和细胞周期途径 有关(q 值 = 2e-28)。包含多种胶原蛋白的两个簇(第 4 组和第 5 组)富含 细胞外基质(ECM)组织和受体功能 (q 值 = 2e-16)(图 5B )。这些结果遵循先前研究的结论,强调了 ECM 基因(尤其是 SPARC 和 COL1A1)与肿瘤侵袭性之间的密切相关性 [ 44 ]。此外,该网络揭示了与 淋巴和非淋巴细胞 通路(第 2 组,q 值 = 6e-10)和 平滑肌收缩 (第 3 组,q 值 = 7e-12)之间的免疫调节。与特征基因结果相似(图 5 A),基因-癌症簇图显示 COAD 和 STAD 在 RNA 共表达方面非常相似(图 5 C),这在早期特征中观察到,并被报道为泛来自癌症组织来源研究的胃肠道癌症(Hoadley 等人,2018 年)。
为了解决患者体内肿瘤异质性的潜在混淆,我们使用 xCell [ 28 ] 对每位患者的细胞类型进行去卷积。然后,我们使用逻辑回归调整所有细胞类型组成的基因,类似于我们之前所做的 [ 44 ]。我们使用 Kendall-Tau 相关评分对两组差异表达 (DE) 基因的细胞类型调整前后进行了比较,类似于之前的研究 [ 29 ]。在 HCC 和 BRCA 上,Kendall-Tau 相关分数分别为 0.52(p 值 < 1.04e-25)和 0.55(p 值 < 3.5e-150)。高度显着的 p 值拒绝了这两个 DE 基因排名是独立的假设。
类似的癌症类型可用作预测模型
受在某些癌症中观察到的相似性的启发,我们探索了这些模型是否适用于迁移学习,也就是说,建立在一种特定癌症类型上的模型可用于预测另一种癌症类型患者的生存率。我们测试了所有对 32 种癌症,交替用作训练和测试数据集。许多癌症模型可有效预测其他癌症类型(图 6 一个)。有趣的是,建立在间皮瘤 (MESO) 数据上的模型显着预测了 12 种其他癌症类型的亚型,长秩 p 值范围从 0.048 到 4.8e-6,C 指数范围从 0.58 到 0.82。一般来说,生物学上更相关的癌症类型具有更高的交叉癌症预测准确度,例如癌症对 COAD/STAD,这是通过早期特征基因分析确定的两种密切的癌症类型(图 5C ) 。STAD 模型显着预测 COAD 样本的亚型(p 值 = 0.018,CI = 0.60)(图 6 B),反之亦然,对于 STAD 样本的 COAD 模型预测(p 值 = 5.4e-3,CI = 0.66 ) (图 6 C)。
由于 READ 和 COAD 之间明显缺乏可预测性(图 5A ),我们进一步调查了潜在来源。READ 显示与 STAD 和 COAD 相似的前 100 个基因表达模式;然而,在 miRNA 和甲基化水平的前 100 个特征中却完全不同(图 5 )。因此,我们仅使用 RNA 作为特征构建了 COAD 和 EAD 的附加模型,并且能够显着提高 COAD 和 READ 之间的相互可预测性(附加文件 4 图 S5)。
讨论
在本报告中,我们提出了一个新颖的通用计算框架,名为 DeepProg,它结合深度学习(自动编码器)和机器学习算法处理多种类型的组学数据集,专门用于生存预测。我们已经展示了 DeepProg 的几个特征,包括其优于其他最先进方法的预测准确性、预测其他 HCC 和 BRCA 人群患者生存的稳健性,以及它作为
迁移学习工具的适用性, 一种相关的癌症来预测另一种癌症
。
DeepProg 的一些独特的机械特性有助于提高其准确性。首先,它使用
增强程序
来增加最终模型的稳健性,方法是聚合来自原始样本不同子集的较弱模型。这种设计非常适合分布式计算架构,并且可以轻松扩展。其次,它对每种组学数据类型都采用了
模块化设计
,并且可以扩展到其他组学和数据类型。在其默认配置中,DeepProg 首先使用
自动编码器单独处理每个组学数据
集,然后在
统一的 Cox-PH 拟合-聚类-监督分类工作流程下合并隐藏层特征
。自编码器结构将各种组学类型的
初始输入特征转换为新特征
。
袋
外样本(internal
out-of-bag
samples)和/或外部数据集。在这项研究中,我们展示了 DeepProg 在整合 3 种组学数据方面的价值:RNA-Seq、microRNA-Seq 和 DNA 甲基化。其他处理
突变
或
病理图像数据
的专用深度学习模型可以作为单独的模块开发并添加到 DeepProg。在预测患者生存率方面,DeepProg 的总体性能明显优于 SNF 等无监督方法。一个主要原因是 SNF 在执行积分时不包含生存信息;相反,它只依赖于来自多种基因组数据的模式。此外,SNF 等无监督方法无法预测 DeepProg 等新样本的预后。
我们进一步使用 DeepProg 来识别 32 种癌症中肿瘤侵袭性的全面特征。尽管之前的几项泛癌研究使用一种或不同的组学类型来了解泛癌分子标志 [
38
,
45
,
46
],这里的报告是第一个系统地描述泛癌生存亚型之间差异的报告。我们确定了与
攻击性亚型
相关的主要生存特征,并专注于 RNA-Seq 表达分析。泛癌基因调控网络突出了这些癌症
最具侵袭性的亚型中最高的共表达基因
。其中许多与
细胞增殖
、
细胞外基质 (ECM) 组织
和
免疫调节
有关,证实了有关癌症侵袭的文献中的早期结果 [
47
,
48
]。这些基因与细胞分裂周期 [
33
]、细胞骨架结构 [
49
]、collagens胶原蛋白 [
48
] 或cadherin families钙粘蛋白家族 [
50 ] 显着相关
]。我们还发现了几个与平滑肌收缩有关的基因。例如,Calponin 基因 CNN1、TAGLN 和 TMP2 在不同的癌症中共表达(图
4
)。此外,CNN1、TAGLN 和 TMP2 已经被定性为膀胱癌的预后分子标志物,其较高的表达与较低的存活率相关 [
51
]。有趣的是,
各种转录因子家族
,如锌指基因被下调,HOX 基因被高甲基化(附加文件
8
:表 S6)。以前的报告支持这种观察,因为多种锌指蛋白已被证明可作为肿瘤抑制基因 [
52
],并且 HOX 基因的失调在癌症中很常见,因为它们中的许多在
细胞分化中
起重要作用 [
53
]。通过实验跟进以测试它们的效果将是有意义的。最后,通过对 32 种癌症的综合比较,揭示了与临床(生存)相关的分子相似性。例如,来自 COAD 和 STAD(两种胃肠癌)的侵袭性亚组呈现出
多种常见模式
。我们推测未来可以利用这些关系来构建更稳健的分析 [
54
],并通过利用患者资料和癌症相似性来帮助制定治疗计划。