-i or --in 指定输入文件,核苷酸序列或者蛋白的fasta序列文件,v5.1.0,输入文件可以指定为目录文件,对文件夹内的fasta进行批量处理;
-o or --out 指定输出结果目录;
-m or --mode:运行模式,可选择genome, proteins, transcriptome;
-c N, --cpu N: Specify the number (N=integer) of threads/cores to use.
-l or --lineage_dataset:指定输入数据库的地址
--offline :配合-l 指定下载的数据库时使用,否则会报错找不到数据库
-f ,--force:Force rewriting of existing files. Must be used when output files with the provided name already exist.
数据库名字例如:bacteria_odb10, 或者 a path i.e. ./bacteria_odb10 or /home/user/bacteria_odb10.
Busco中建议使用前一个示例引入数据库文件,Busco会自动下载引入的数据库文件;后者示例中,从指定的路径文件中寻找数据库文件。如果使用自动的lineage,Lineage会被忽略;
Lineage can be ignored if running automated lineage selection.
config files:
In the config/ subfolder of the cloned repository, a config.ini template is provided. In this file, you may declare the paths to all third party components matching what is on your machine. To activate this config file, set the environment variable BUSCO_CONFIG_FILE with the path to the file, as follows:
export BUSCO_CONFIG_FILE=“/path/to/myconfig.ini”
Alternatively you may pass the path to your config file by using the --config /path/to/config.ini command line option. This is useful for switching between configurations or manage parameters for each run in a dedicated file.
运行了4h~,我展示了一下某物种参考基因组的busco结果:
最主要的结果在short_summary…2018.txt中,
C:多少个BUSCO测试基因被覆盖,C=S+D;
S:多少个基因经过比对发现是单拷贝;
D:多少个基因经过比对发现包含多拷贝;
F:多少个基因经过比对覆盖不完全,只是部分比对上;
M:没有得到比对结果的基因数;
Total:总共测试的基因条目数,Total=C+F+M。
此外,还可以进行多物种的busco结果的比较,这个就先不展示了。
五.参考:
https://www.sohu.com/a/213046854_464200
https://busco.ezlab.org/
https://www.jianshu.com/p/0ed311feaffa
文献引用:
The novelties introduced in BUSCO v4 and v5 and the new BUSCO datasets (*_odb10) are described here.If you’ve used these versions the correct citation would be:
Mosè Manni, Matthew R Berkeley, Mathieu Seppey, Felipe A Simão, Evgeny M Zdobnov, BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Molecular Biology and Evolution, Volume 38, Issue 10, October 2021, Pages 4647–4654
Additional protocols and applications are described in: Manni, M., Berkeley, M. R., Seppey, M., & Zdobnov, E. M. (2021). BUSCO: Assessing genomic data quality and beyond. Current Protocols, 1, e323. doi: 10.1002/cpz1.323
隔壁的室友
Fluster是一个平台,旨在通过使用智能功能简化对室友和公寓的搜索 :grinning_face_with_big_eyes: :party_popper:
这个的后端,由 , 和构建。
Fluster提供的一些功能:
Fluster推荐适合您需求的公寓
有了Fluster,您可以发现未来室友的生活方式和爱好
您喜欢Fluster上的一个地方吗? 您可以发送即时查看请求
Fluster会照顾您的观看日历
Fluster是免费的,开源的,甚至发布有关您房间的广告,让它也免费
随时向我发送有关反馈或。 始终欢迎功能要求。
在或关注Fluster。
Fluster由David Dal Busco开发。
Fluster徽标是瑞士苏黎世David Dal Busco的注册商标。 如果要使用它,请与联系。
这是一个用于评估常见基因组装配质量参数的Python程序。
由托马斯·尼尔森(Thomas Nelson)撰写。
此脚本需要Biopython模块和Python 3.4或更高版本。
在此处获取Biopython模块: ://biopython.org/wiki/Download。
用法:python assemblyQC.py [包含程序集的FASTA文件名]
该程序将在使用标准服务器类Linux盒的2.5 Gbp基因组装配上在一两分钟内返回结果。
QUAST执行快速方便的质量评估和基因组装配比较。 QUAST计算了许多众所周知的指标,包括重叠群准确性,发现的基因数量,N50和其他指标,以及引入了新的指标,例如NA50(请参见本文和手册中的详细信息)。 全面的分析得出汇总表(纯文本,制表符分隔和LaTeX格式)和彩色图表。 该工具还可以生成基于Web的报告,将所有信息压缩到一个易于浏览的文件中。 QUAST具有直观的命令行界面和详细的手册,可帮助用户运行它并了解其输出。 此外,实验室在http://quast.bioinf.spbau.ru/上启动了Web-QUAST的Beta版,这使质量评估更加简单。 QUAST和MetaQUAST(宏基因组程序集的扩展名)论文发表在《生物信息学》上。
BUSCO目前已经更新到第4版,之前只用来评估基因组为目标,现在还能够预测基因和做系统基因组学分析。
官方提供了docker,conda和GitLab这三种方法,这里我只介绍conda。
conda create -n busco4 -c bioconda -c conda-forge busco=4.0.5
conda activate busco4
由于软件运行的时...
点击上面“蓝字”关注我们!生 信 老 司 机 教 你如何做基因组项目转眼间,从事生信工作已数年有余。在这期间,一直专注于基因组方面。项目经验较多,涉及的物种也从微生物扩展到动植物,但其中不乏有各种奇怪的项目,遇到各样匪夷所思的问题,也难免有不甚理想的结果。从中既是体会到了成长的痛苦,也感受过更多开花结果的喜悦。猛然间,从内心流露出一丝希冀,想要证明自己在所爱的路上,曾经努力过,也终有所...
基因组组装
基因组组装一般分为三个层次,contig, scaffold和chromosomes. contig表示从大规模测序得到的短读(reads)中找到的一致性序列。组装的第一步就是从短片段(pair-end)文库中组装出contig。进一步基于不同长度的大片段(mate-pair)文库,将原本孤立的c...
rust-mdbg 是一种超快的minimizer-space de Bruijn graphs (mdBG) 实现,适用于组装长而准确的读数,例如PacBio HiFi。
随着18年以来Pacbio HiFi reads的出现,让一些复杂基因组的组装不再复杂,而且有越来越多的课题组也加入到了基因组学的研究中,正是因为有了高精度长读长的reads,目前也产生了很多专门用于HiFi组装的软件,如Hifiasm,当然这篇文章的软件的算法,可以用超短时间,低内存去组装。我相信随着不断的发展,以后做组装的
琪丁大圣:动态的相亲网页,效果做的不错。特别是搜索功能,能把数据库的数据导出来;
何广强:恩,怎么说呢,其实也挺不错,但是有点离题了;
等灯等灯:网页布局什么的都很好,如果把用户的信息等连到数据库,那这个网址实用性又增强了不少;
打的是自己人:app演示的炫好棒,如果能程序自动输出题目就好了。
大哥特工队:app界面及功能冲旷了我们的视野。
雷锋队:这个网页真的做的很吊!居然是用二维码扫...
先附上蜗蜗科技前辈对于这俩名词的理解:http://www.wowotech.net/armv8a_arch/atomicity.html
我把这个博客看完了,armv8手册上也有相关解释,我不想看,可读性太差。
这里说一下个人理解:
1、single copy的意思是core连续多次对同一地址发起load或者store操作,原子性保证的单位是byte,那么就是说这个byte
参考:【干货】基因组组装你了解多少? -- 诺禾致源 动植物基因组de novo工作,其组装指标的好坏直接影响着整个基因组的质量。而评估基因组组装结果,contigN50和scaffoldN50是第一指标,即contig/ scaffoldN50:将contig/scaffold长度从长到短进行排序并累加,当累加和达到contig/scaffold总长度的50%的时候,最后参与加和的那一条...
quast 的结果怎么看The assembly algorithms that have been developed so far intend to provide better assemblies evaluated under different criteria. Hence, depending on the specific scenario the assembly proce...
宏基因组数据进行分箱(binning)后,需要对得到的宏基因组组装基因组(MAG)进行质量评估。常用的工具是CheckM,主要以每个MAG的completeness与contamination来作为判断指标。
Bowers, R., Kyrpides, N., Stepanauskas, R. et al. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG)


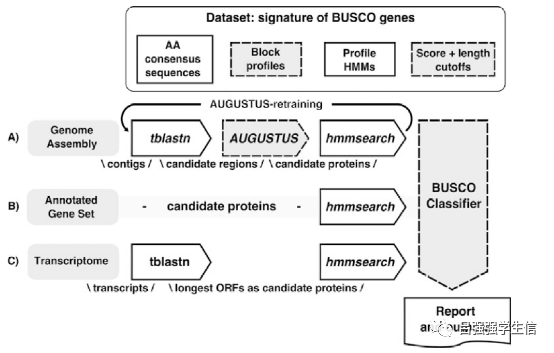 BUSCO流程框架图
BUSCO流程框架图